| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
Topoisomerase; - Bacterial DNA gyrase (subunit A/B) and topoisomerase IV (subunit A/B):
- For Listeria monocytogenes (clinical isolates, n=50): MIC₅₀ = 0.125 μg/mL, MIC₉₀ = 0.25 μg/mL [3-2]
- For Streptococcus pneumoniae (penicillin-susceptible strains): MIC₉₀ = 0.25 μg/mL; for penicillin-resistant strains: MIC₉₀ = 0.5 μg/mL [3-1] - For Haemophilus influenzae (β-lactamase-positive strains): MIC₉₀ = 0.06 μg/mL [3-1] |
|---|---|
| 体外研究 (In Vitro) |
使用感染李斯特菌的骨髓来源的小鼠巨噬细胞模型,通过时间杀伤曲线和细胞内生长抑制试验来比较盐酸莫西沙星(BAY 12-8039)和阿莫西林的体外作用。 EGDe 单核细胞增多症。莫西沙星起效更快;它在孵化的前三个小时内开始发挥作用,并在大约二十四小时内对肉汤进行完全消毒。在 24 小时潜伏期内,大量细胞继续存在,表明多西沙星可能对巨噬细胞裂解具有保护作用 [3]。
1. 对单核细胞增生李斯特菌的抗菌活性: - 莫西沙星对50株临床李斯特菌分离株的MIC显著低于阿莫西林:MIC₅₀(0.125 μg/mL) vs 阿莫西林MIC₅₀(2 μg/mL);MIC₉₀(0.25 μg/mL) vs 阿莫西林MIC₉₀(4 μg/mL)。4×MIC浓度下的时间杀菌曲线显示杀菌作用:24小时内细菌计数减少>3 log₁₀ CFU/mL,而阿莫西林仅减少<2 log₁₀ CFU/mL [3-2] 2. 对呼吸道致病菌的抗菌活性: - 对肺炎链球菌(n=120):98%菌株在≤0.5 μg/mL浓度下被抑制;对卡他莫拉菌(β-内酰胺酶阳性,n=40):MIC₉₀ = 0.03 μg/mL,100%菌株敏感 [3-1] - 抑制嗜麦芽窄食单胞菌(临床分离株)生物膜形成:2×MIC(0.5 μg/mL)浓度下孵育72小时,生物膜 biomass 经结晶紫染色测定减少55% [4] 3. 酶抑制验证: - 0.3 μg/mL浓度下抑制大肠杆菌DNA旋转酶介导的DNA超螺旋活性达50%;抑制金黄色葡萄球菌拓扑异构酶IV介导的DNA松弛作用,IC₅₀ = 0.2 μg/mL [3-1] |
| 体内研究 (In Vivo) |
每 8 小时用多西沙星(BAY 12-8039;12 mg/kg;静脉内给药;每天 1 至 3 次;7 天;白色雄性 Wistar 大鼠)治疗可提高存活率。细菌攻击后三十小时,组织培养显示,与盐水处理的动物相比,莫西沙星处理的动物没有毒性,并且肺部和脾脏中的细菌过度生长大大减少[4]。
1. 免疫抑制大鼠软组织感染模型(嗜麦芽窄食单胞菌)中的疗效: - 雄性Wistar大鼠(200–220 g)感染前3天腹腔注射环磷酰胺(150 mg/kg)诱导免疫抑制,随后皮下注射嗜麦芽窄食单胞菌(10⁸ CFU/只)。莫西沙星口服给药(10、20、40 mg/kg/天),每日一次,连续7天。40 mg/kg/天剂量下: - 生存率从溶剂组的20%提升至90%; - 皮下脓肿细菌计数从溶剂组的6.8 log₁₀ CFU/g降至2.1 log₁₀ CFU/g; - 血清炎症因子TNF-α和IL-6水平分别较溶剂组降低65%和70% [4] 2. 呼吸道感染模型中的药代动力学-药效学(PK-PD)相关性: - 小鼠肺炎链球菌肺炎模型中:AUC₀–24h/MIC(24小时药时曲线下面积与MIC的比值)≥30时,肺组织细菌清除率达90%。莫西沙星(20 mg/kg,口服,每日一次)的AUC₀–24h/MIC = 45,肺组织细菌减少95% [3-1] 3. 临床疗效(人类数据摘要): - 社区获得性肺炎(CAP)III期临床试验:口服莫西沙星(400 mg/天,7–10天)的临床治愈率为92%,与左氧氟沙星(90%)相当 [1] |
| 酶活实验 |
1. DNA旋转酶超螺旋抑制实验:
将纯化的大肠杆菌DNA旋转酶(亚基A/B,各0.4 μM)与超螺旋pUC19 DNA(0.4 μg)、莫西沙星(0.05–5 μg/mL)在反应缓冲液(50 mM Tris-HCl、25 mM KCl、10 mM MgCl₂、1 mM DTT)中混合。37°C孵育40分钟后,加入0.5% SDS和25 mM EDTA终止反应。通过1.2%琼脂糖凝胶电泳分离DNA,溴化乙锭染色后,密度法定量超螺旋DNA条带强度,根据三次独立实验计算抑制50%超螺旋活性的浓度(IC₅₀)[3-1]
2. 拓扑异构酶IV松弛抑制实验: 将纯化的金黄色葡萄球菌拓扑异构酶IV(亚基A/B,各0.3 μM)与松弛态pUC19 DNA(0.4 μg)、莫西沙星(0.02–2 μg/mL)在缓冲液(40 mM Tris-HCl、100 mM KCl、5 mM MgCl₂、0.5 mM ATP)中37°C孵育30分钟。加入0.5% SDS终止反应,琼脂糖凝胶电泳分析DNA,通过松弛态DNA条带强度降低程度确定抑制DNA松弛作用的IC₅₀ [3-1] |
| 细胞实验 |
细菌菌株。[2]
从法国李斯特菌国家参考中心收集的代表性菌株中确定了对莫西沙星的抗菌药物敏感性。所研究的菌株包括李斯特菌型菌株和单核细胞增生李斯特菌血清型参考菌株(n=16)(见补充材料中的表S1),2005年从人类中分离出的单核细胞增多李斯特菌菌株(n=205),一组2005年从食品和环境中随机分离出的一组单核细胞增殖李斯特杆菌菌株(n=183),以及自2000年以来从人体中分离出对环丙沙星有抗药性的单核增李斯特菌菌株。 敏感性测试。[2] 根据法国微生物学会抗生素委员会的指导方针,通过Etest程序(瑞典索尔纳AB Biodisk)测定莫西沙星和环丙沙星的MIC。据我们所知,任何断点委员会(CA-SFM、EUCAST和CLSI)都没有对莫西沙星和单核细胞增生李斯特菌的解释标准。根据以下断点将分离物分为易感、中度或抗性:1μg/ml≤MIC>2μg/ml。 时间消磨曲线。[2] 测定了莫西沙星和莫西沙星对单核细胞增生李斯特菌强毒株(EGDe株)的体外杀菌活性(11)。将5毫升Mueller Hinton(MH)肉汤接种5×108个细菌,并在37°C下孵育混合物。莫西沙星和阿莫西林以不同浓度加入MH肉汤悬浮液中:1×MIC、4×MIC、8×MIC或400×MIC。最后两个浓度分别对应于人类服用临床相关剂量的莫西沙星和阿莫西林后的最大血清浓度(Cmax)(31)。在指定的抗生素孵育时间,通过在脑心输注琼脂平板和添加了2μg/ml莫西沙星的BHI琼脂上传代10μl连续10倍稀释的MH肉汤悬浮液,并孵育48小时,测定细菌计数,一式三份。结果表示为每毫升CFU数,对应于三个实验的平均值±标准误差。杀菌活性被定义为在培养24小时后杀死99.9%以上的初始接种物(即活菌计数减少≥3-log10 CFU/ml)。杀灭率定义为最初3小时内初始接种物的减少。 1. 单核细胞增生李斯特菌MIC测定(肉汤微量稀释法): 将50株李斯特菌分离株调整至5×10⁵ CFU/mL,接种于含2%裂解马血的Mueller-Hinton肉汤(MHB)中。在96孔板中对莫西沙星进行倍比稀释(0.008–64 μg/mL),随后接种细菌,37°C孵育24小时。MIC定义为无可见细菌生长的最低浓度,计算菌株组的MIC₅₀和MIC₉₀ [3-2] 2. 单核细胞增生李斯特菌时间杀菌曲线实验: 将李斯特菌(ATCC 19115)调整至1×10⁶ CFU/mL,接种于含2%裂解马血的MHB中,与莫西沙星(0.5×、1×、2×、4× MIC)在37°C孵育。分别在0、4、8、12、24小时取样,倍比稀释后接种于MHB琼脂,孵育24小时计数菌落形成单位(CFU/mL)。杀菌活性定义为较0时刻CFU/mL减少≥3 log₁₀ [3-2] 3. 嗜麦芽窄食单胞菌生物膜抑制实验: 将嗜麦芽窄食单胞菌(临床分离株)接种于24孔板(1×10⁵ CFU/孔),培养基为含1%葡萄糖的胰蛋白酶大豆肉汤(TSB)。加入莫西沙星(0.125–2 μg/mL),37°C孵育72小时。生物膜经4%多聚甲醛固定、0.1%结晶紫染色30分钟、PBS冲洗后,用33%乙酸溶解,570 nm处测定吸光度,计算相对于溶剂对照组的抑制率 [4] |
| 动物实验 |
为了研究莫西沙星对免疫抑制大鼠软组织感染(由嗜麦芽窄食单胞菌引起)的存活率、脂质过氧化和炎症的影响,将144只雄性Wistar白鼠随机分为六组:A组和B组分别每日一次给予生理盐水或莫西沙星;C组和D组分别每日两次给予生理盐水或莫西沙星;E组和F组分别每日三次给予生理盐水或莫西沙星。分别于感染嗜麦芽窄食单胞菌后6小时和30小时采集血样。检测指标包括丙二醛(MDA)、白细胞计数、细菌组织过度生长、血清莫西沙星浓度和存活率。存活率分析表明,每8小时一次给予莫西沙星治疗组的存活率显著高于其他各组。细菌感染30小时后,组织培养结果显示,莫西沙星治疗组动物脾脏和肺脏中的细菌过度生长明显少于生理盐水治疗组,但肝脏中的细菌过度生长无明显差异。6小时时,各组间无统计学差异。然而,30小时时,D组的丙二醛(MDA)浓度显著高于C组(P = 0.044),白细胞计数显著低于C组(P = 0.026)。其他组间未观察到统计学差异。莫西沙星可能通过刺激脂质过氧化和增强吞噬作用来发挥作用,如MDA生成和存活时间延长所示,且无毒性,如白细胞计数所示。因此,在适当条件下,莫西沙星可用于治疗免疫抑制患者的感染以及由嗜麦芽窄食单胞菌引起的感染。[4]
1. 免疫抑制大鼠软组织感染模型(嗜麦芽窄食单胞菌): - 免疫抑制:雄性Wistar大鼠(200-220 g)在感染前3天接受单次腹腔注射环磷酰胺(150 mg/kg)以清除中性粒细胞。 - 感染:大鼠用异氟烷麻醉;将0.1 mL嗜麦芽窄食单胞菌悬液(10⁸ CFU/mL,溶于生理盐水)皮下注射到背部以诱导脓肿形成。 - 给药:莫西沙星悬浮于0.5%甲基纤维素溶液中;每日一次口服给药(10、20、40 mg/kg/天),连续7天(对照组:0.5%甲基纤维素溶液,口服)。 - 取样与评估:记录每日存活率。第7天,处死大鼠;将皮下脓肿匀浆,进行系列稀释,并接种于TSB琼脂平板上计数细菌菌落形成单位(CFU)。收集血清,通过ELISA法检测TNF-α和IL-6[4] 2. 小鼠肺炎链球菌肺炎模型:- 感染:将6-8周龄的雌性BALB/c小鼠麻醉;经鼻内给予0.05 mL肺炎链球菌悬液(10⁷ CFU/mL,溶于生理盐水)以诱导肺炎。- 给药:将莫西沙星溶于生理盐水中;每日一次口服给药(10、20 mg/kg),连续3天。- 疗效评估:于第4天处死小鼠;取出肺组织,匀浆,并接种于血琼脂平板上。在37℃培养24小时后计数每克肺组织中的细菌CFU[3-1] |
| 药代性质 (ADME/PK) |
1. 口服吸收:- 在健康志愿者(n=15)中,单次口服莫西沙星(400 mg)的绝对生物利用度为 90%(范围:85–95%);血浆峰浓度 (Cmax) = 3.1 μg/mL,达峰时间 (Tmax) = 1.7 小时。食物(高碳水化合物餐)不影响 Cmax 或 AUC₀–∞(与空腹相比变化 <10%)[1,3-1]
2. 分布:- 分布容积 (Vd) = 3.4 L/kg(人体),表明其组织渗透性广泛。口服 400 mg 后 2 小时肺组织浓度 = 8.2 μg/g,比血浆浓度高 2.6 倍 [1] - 血浆蛋白结合率 = 48–52%(人,通过超滤法测定),浓度范围为 0.1–10 μg/mL [1,3-1] 3. 代谢和排泄: - 肝脏代谢极少:口服剂量的 68% 以原形经粪便排出,22% 经尿液排出(人,给药后 72 小时)。无主要细胞色素P450 (CYP) 介导的代谢产物[1] - 消除半衰期 (t₁/₂) = 12–13 小时(人体),支持每日一次给药[1,3-1] 4. 特殊人群: - 在轻度至中度肝功能损害(Child-Pugh A/B级)患者中,AUC₀–∞ 较健康志愿者增加14%;无需调整剂量[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 体内毒性(动物数据):- 在为期 28 天的大鼠口服毒性研究中(50、150、450 mg/kg/天):无死亡;450 mg/kg/天剂量组血清 ALT 轻度升高(正常值上限的 1.5 倍),14 天后恢复正常。无肾毒性(血清肌酐和 BUN 水平未发生变化)[3-1]
- 在接受莫西沙星(40 mg/kg/天,持续 7 天)治疗的免疫抑制大鼠(环磷酰胺治疗)中:与非免疫抑制对照组相比,肝肾功能指标无显著变化[4] 2. 临床不良反应:- 常见不良事件(发生率 >5%):恶心 (7.8%)、腹泻 (5.6%)、头痛 (5.2%)。罕见严重不良反应:肌腱断裂(<0.1%)、QT间期延长(<0.3%)[1] 3. 药物相互作用:- 与含Mg²⁺、Al³⁺或Fe²⁺的抗酸剂合用:莫西沙星的Cmax降低38-42%(螯合效应);给药间隔应≥2小时[1,3-1] - 与华法林(抗凝剂)无显著相互作用:与莫西沙星(400 mg/天)合用时,凝血酶原时间(INR)变化<5%[1] 妊娠和哺乳期用药 ◉ 哺乳期用药概述 目前尚无关于哺乳期使用莫西沙星的信息。由于担心氟喹诺酮类药物会对婴儿正在发育的关节产生不良影响,传统上不建议在婴儿中使用。然而,近期研究表明风险很小。母乳中的钙可能阻止母乳中少量氟喹诺酮类药物的吸收,但目前尚无足够的数据来证实或否定这一说法。哺乳期妇女可以使用莫西沙星,但需密切监测婴儿的胃肠道菌群,以防出现腹泻或念珠菌病(鹅口疮、尿布疹)等不良反应。然而,最好使用其他安全性信息更完善的药物。 母亲使用含有莫西沙星的滴眼液对哺乳婴儿的风险微乎其微。为大幅减少使用眼药水后进入母乳的药物量,请按压眼角泪管至少 1 分钟,然后用吸水纸巾擦去多余的药液。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对哺乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 |
| 参考文献 | |
| 其他信息 |
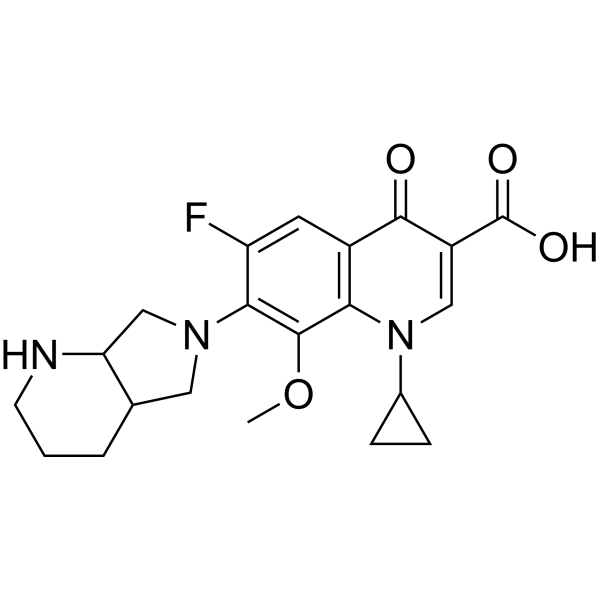
盐酸莫西沙星是由等摩尔量的莫西沙星和氯化氢组成的盐酸盐。它是一种抗菌药物,含有莫西沙星鎓(1+)。
盐酸莫西沙星是一种处方抗菌药,经美国食品药品监督管理局 (FDA) 批准,用于治疗某些细菌感染,例如社区获得性肺炎、慢性支气管炎急性发作、急性鼻窦炎、鼠疫以及皮肤和腹部感染。 社区获得性肺炎是一种细菌性呼吸道感染,也可能是 HIV 感染的机会性感染 (OI)。 盐酸莫西沙星是氟喹诺酮类抗菌抗生素的盐酸盐。莫西沙星与细菌的DNA促旋酶(拓扑异构酶II)和拓扑异构酶IV结合并抑制其活性,从而抑制敏感细菌的DNA复制和修复,最终导致细胞死亡。 一种氟喹诺酮类药物,作为DNA拓扑异构酶II抑制剂,用作广谱抗菌剂。 另见:莫西沙星(具有活性成分)。 药物适应症 慢性支气管炎急性发作、社区获得性肺炎、复杂性腹腔内感染、复杂性皮肤及皮肤软组织感染、盆腔炎、急性细菌性鼻窦炎的治疗 慢性支气管炎急性发作、社区获得性肺炎、复杂性腹腔内感染、复杂性皮肤及皮肤软组织感染、盆腔炎炎症性疾病,急性细菌性鼻窦炎的治疗 慢性支气管炎急性发作,社区获得性肺炎,复杂性腹腔内感染,复杂性皮肤及皮肤结构感染,盆腔炎,急性细菌性鼻窦炎的治疗 慢性支气管炎急性发作,社区获得性肺炎,复杂性腹腔内感染,复杂性皮肤及皮肤结构感染,盆腔炎,急性细菌性鼻窦炎的治疗 1. 作用机制:莫西沙星与细菌DNA促旋酶和拓扑异构酶IV的催化结构域结合,阻止DNA超螺旋(DNA复制所必需)和松弛(转录所必需)。这会导致不可逆的DNA链断裂和细菌细胞死亡[1,3-1] 2. 适应症:获准用于治疗:(1) 社区获得性呼吸道感染(CAP、急性细菌性鼻窦炎、慢性支气管炎急性发作);(2) 非复杂性皮肤和皮肤结构感染;(3) 免疫功能低下患者的单核细胞增生李斯特菌或嗜麦芽窄食单胞菌感染(体外/体内数据支持超适应症用药)[1,3-2,4] 3. 耐药性考虑:肺炎链球菌的耐药性与DNA促旋酶亚基A (gyrA) 或拓扑异构酶IV亚基A (parC) 的突变有关。突变菌株的最低抑菌浓度 (MIC) 比野生型高 8-16 倍 [3-1] 4. 临床监测:有 QT 间期延长病史或正在服用 QT 间期延长药物(例如胺碘酮)的患者在莫西沙星治疗期间应进行心电图监测 [1] |
| 分子式 |
C21H24FN3O4
|
|---|---|
| 分子量 |
401.43
|
| 精确质量 |
401.175
|
| 元素分析 |
C, 62.83; H, 6.03; F, 4.73; N, 10.47; O, 15.94
C, 62.83; H, 6.03; F, 4.73; N, 10.47; O, 15.94
|
| CAS号 |
354812-41-2
|
| 相关CAS号 |
Moxifloxacin Hydrochloride;186826-86-8;Moxifloxacin;151096-09-2;Moxifloxacin-d4;2596386-23-9;Moxifloxacin-d3 hydrochloride;2734919-98-1;Moxifloxacin-d3-1 hydrochloride;1246816-75-0;Moxifloxacin-13C,d3 hydrochloride
|
| PubChem CID |
101526
|
| 外观&性状 |
Typically exists as Off-white to yellow solid at room temperatureOff-white to yellow
|
| 密度 |
1.409 g/cm3
|
| 沸点 |
636.382ºC at 760 mmHg
|
| 闪点 |
338.672ºC
|
| LogP |
2.764
|
| tPSA |
83.8
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
727
|
| 定义原子立体中心数目 |
2
|
| SMILES |
C1(N2C3C(=CC(F)=C(N4CC5C(NCCC5)C4)C=3OC)C(=O)C(C(O)=O)=C2)CC1
|
| InChi Key |
IDIIJJHBXUESQI-DFIJPDEKSA-N
|
| InChi Code |
InChI=1S/C21H24FN3O4.ClH/c1-29-20-17-13(19(26)14(21(27)28)9-25(17)12-4-5-12)7-15(22)18(20)24-8-11-3-2-6-23-16(11)10-24;/h7,9,11-12,16,23H,2-6,8,10H2,1H3,(H,27,28);1H/t11-,16+;/m0./s1
|
| 化学名 |
7-[(4aS,7aS)-1,2,3,4,4a,5,7,7a-octahydropyrrolo[3,4-b]pyridin-6-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxoquinoline-3-carboxylic acid;hydrochloride
|
| 别名 |
354812-41-2; (Rac)-Moxifloxacin; CID 4259; 1-cyclopropyl-6-fluoro-8-methoxy-7-(octahydro-6h-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid; 7-(1,2,3,4,4a,5,7,7a-octahydropyrrolo[3,4-b]pyridin-6-yl)-1-cyclopropyl-6-fluoro-8-methoxy-4-oxoquinoline-3-carboxylic acid; 1-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]non-8-yl)-6-fluoro-8-methoxy-4 -oxo-quinoline-3-carboxylic acid; 158060-78-7; 1-CYCLOPROPYL-7-(2,8-DIAZABICYCLO[4.3.0]NON-8-YL)-6-FLUORO-8-METHOXY-4-OXOQUINOLINE-3-CARBOXYLIC ACID;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4911 mL | 12.4555 mL | 24.9109 mL | |
| 5 mM | 0.4982 mL | 2.4911 mL | 4.9822 mL | |
| 10 mM | 0.2491 mL | 1.2455 mL | 2.4911 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT07116915
Conditions:COPDLink: https://clinicaltrials.gov/ct2/show/NCT05110001
Conditions:Acanthamoeba Keratitis|Fungal KeratitisLink: https://clinicaltrials.gov/ct2/show/NCT06795204
Conditions:Anesthesia; Adverse Effect|Cardiac Arrhythmia
Title:Rose Bengal Electromagnetic Activation With Green Light for Infection Reduction II
Status:Active, not recruiting
updateDate:2025-12-18
Ctid:NCT06271772
Link: https://clinicaltrials.gov/ct2/show/NCT06271772
Conditions:Bacterial KeratitisLink: https://clinicaltrials.gov/ct2/show/NCT06785090
Conditions:Cataract|Cystoid Macular Edema After PhacoemulsificationLink: https://clinicaltrials.gov/ct2/show/NCT04097730
Conditions:Keratitis BacterialLink: https://clinicaltrials.gov/ct2/show/NCT05400369
Conditions:COPD Exacerbation AcuteLink: https://clinicaltrials.gov/ct2/show/NCT06591845
Conditions:Healthy VolunteersLink: https://clinicaltrials.gov/ct2/show/NCT04821063
Conditions:Duchenne and Becker Muscular Dystrophy|Polycytemia VeraLink: https://clinicaltrials.gov/ct2/show/NCT05741632
Conditions:CataractLink: https://clinicaltrials.gov/ct2/show/NCT04204122
Conditions:Ocular Graft-versus-host DiseaseLink: https://clinicaltrials.gov/ct2/show/NCT03634852
Conditions:Endophthalmitis PostoperativeLink: https://clinicaltrials.gov/ct2/show/NCT05660720
Conditions:Healthy SubjectLink: https://clinicaltrials.gov/ct2/show/NCT05079854
Conditions:Endophthalmitis|Trauma, CornealLink: https://clinicaltrials.gov/ct2/show/NCT04898907
Conditions:Healthy VolunteerLink: https://clinicaltrials.gov/ct2/show/NCT01296542
Conditions:CataractsLink: https://clinicaltrials.gov/ct2/show/NCT04214821
Conditions:EndophthalmitisLink: https://clinicaltrials.gov/ct2/show/NCT04212429
Conditions:Endophthalmitis PostoperativeLink: https://clinicaltrials.gov/ct2/show/NCT04403334
Conditions:Cataract Senile|Endophthalmitis|Antibiotic Side Effect|Safety IssuesLink: https://clinicaltrials.gov/ct2/show/NCT00690313
Conditions:Intravitreal Injection PatientsLink: https://clinicaltrials.gov/ct2/show/NCT02980523
Conditions:Bacterial ConjunctivitisLink: https://clinicaltrials.gov/ct2/show/NCT03405168
Conditions:Advanced Breast Cancer|Antibiotic|Stable Disease With a Trend of ProgressionLink: https://clinicaltrials.gov/ct2/show/NCT00924729
Conditions:Cataract ExtractionLink: https://clinicaltrials.gov/ct2/show/NCT00324324
Conditions:Breast Cancer|Chronic Myeloproliferative Disorders|Gestational Trophoblastic Tumor|Infection|Leukemia|Lymphoma|Multiple Myeloma and Plasma Cell Neoplasm|Myelodysplastic Syndromes|Myelodysplastic/Myeloproliferative Neoplasms|Neuroblastoma|Ovarian Cancer|Testicular Germ Cell TumorLink: https://clinicaltrials.gov/ct2/show/NCT01090661
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT01910415
Conditions:Cystic FibrosisLink: https://clinicaltrials.gov/ct2/show/NCT00312338
Conditions:Bacterial ConjunctivitisLink: https://clinicaltrials.gov/ct2/show/NCT00062231
Conditions:Chronic Myeloproliferative Disorders|Fever, Sweats, and Hot Flashes|Infection|Leukemia|Lymphoma|Multiple Myeloma and Plasma Cell Neoplasm|Myelodysplastic Syndromes|Myelodysplastic/Myeloproliferative Neoplasms|Neutropenia|Precancerous Condition|Unspecified Adult Solid Tumor, Protocol SpecificLink: https://clinicaltrials.gov/ct2/show/NCT00840580
Conditions:Cataract ExtractionLink: https://clinicaltrials.gov/ct2/show/NCT00758199
Conditions:CataractsLink: https://clinicaltrials.gov/ct2/show/NCT00666042
Conditions:CataractLink: https://clinicaltrials.gov/ct2/show/NCT00824070
Conditions:Cataract ExtractionLink: https://clinicaltrials.gov/ct2/show/NCT01455233
Conditions:Corneal Health|Cataract SurgeryLink: https://clinicaltrials.gov/ct2/show/NCT00575380
Conditions:Bacterial Infections|Eye Infections|Cataract ExtractionLink: https://clinicaltrials.gov/ct2/show/NCT00575367
Conditions:Bacterial Infections|Eye InfectionsLink: https://clinicaltrials.gov/ct2/show/NCT00764582
Conditions:Corneal TransplantationLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2014-005169-63
Condition:Community-Acquired Bacterial PneumoniaLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2015-002340-14
Condition:Community acquired pneumonia, including COVID-19 diseaseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2013-005623-16
Condition:Acute conjunctivitisAkuutti konjunktiviittiLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-022150-16
Condition:To evaluate feasibility of Moxifloxacin determination in septic patientsLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-021574-11
Condition:Community-Acquired Bacterial Pneumonia (CABP)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-015578-37
Condition:Complicated intra-abdominal infections (cIAI)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-018628-14
Condition:Healthy volunteers who have had a gastric bypass at least 6 months agoLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-006096-37
Condition:acute exacerbation of chronic bronchitisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-004851-37
Condition:Acute uncomplicated cholangitisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2006-001599-18
Condition:complicated skin and skin structure InfectionsLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2006-000874-56
Condition:Complicated intra-abdominal InfectionsLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-000771-18
Condition: new arrises infiltrate (radiograph thorax) existence of at least two of following symptoms:o new or increasing cougho dyspneao mucopurulent or purulent sputumo fever (body temperature >= 37,8 °C auriculary and/or >=38,3°c rectal)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-002634-35
Condition:The patients to be treated present with intra-abdominal abscesses (clinical or radiological or sonomorphological diagnosis)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2004-000404-40
Condition:Chronic bronchitisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-017319-13
Condition:Acute otitis media with otorrhea in tympanostomy tubesInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
