| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
PARP1 and 2; tubulin/microtubule; tubulin polymerization
|
|---|---|
| 体外研究 (In Vitro) |
AMXI-5001有效且选择性地抑制PARP1和PARP2。AMXI-5001与人PARP1的催化结构域结合。AMXII-5001是一种周糖醛酸酶抑制剂。AMXI-5001是一种强效的微管蛋白聚合抑制剂。AMXI-5001没有明显的脱靶效应。AMXI-5001靶向具有或不具有同源重组缺陷的肿瘤细胞。
AMXI-5001对细胞检查点、调节蛋白和信号蛋白的影响。
AMXI-5001与体外批准的抗癌疗法的协同抗癌活性[1]。
|
| 体内研究 (In Vivo) |
MDA-MB-436异种移植物的初步5天研究[1]
AMXI-5001(HCl盐形式)配制于10%TPGS中,口服给药于携带已建立的MDA-MB-436异种移植物肿瘤的雌性无胸腺裸鼠,MDA-MB-434是一种具有BRCA1缺失和BRCA1缺陷的侵袭性基底乳腺癌细胞系。当肿瘤达到350mm3的平均体积时,以12.5、25或50mg/kg/剂BID的剂量口服AMXI-5001混悬液5天。与赋形剂组相比,每天两次(BID)口服AMXI-5001五天,在25和50 mg/kg/剂BID的剂量下,肿瘤生长受到快速和显著的抑制,在50 mg/kg/剂量BID时,肿瘤明显消退(图S32A)。 此外,血浆和肿瘤组织生物分析显示,AMXI-5001血浆浓度呈剂量依赖性增加,AMXI-001肿瘤浓度也呈相应的剂量依赖性增长(图S32B)。然而,在第4次和第10次重复口服剂量后收获的组织之间,血浆或肿瘤组织中的AMXI-5001浓度没有显著变化(图S32B)。这些结果表明,重复治疗后,AMXI-5001在血液或肿瘤组织中的积聚没有显著增加。 肿瘤裂解物的Western Blot分析表明,AMXI-5001以剂量依赖的方式在体内调节其靶点(图S33)。特别是,AMXI-5001对其靶点的抑制在25和50 mg/kg BID剂量下更为明显,如PARP活性的替代标记物聚合二磷酸腺苷(ADP)核糖(标准杆数)表达显著降低,以及微管聚合抑制的替代物总微管蛋白表达显著降低(图S33)。这为AMXI-5001的抗肿瘤功效可能源于抑制PARP和微管聚合的假设提供了支持。一般来说,在测试剂量下,血浆和肿瘤药物浓度的增加与AMXI-5001靶点之一或两者的抑制增加之间存在良好的相关性。 MDA-MB-436异种移植物的31天验证性研究[1] 在为期5天的研究中观察到的AMXI-5001的疗效在随后的更长给药时间的研究中得到了证实。通过在雌性无胸腺裸鼠的第三乳脂肪中皮下接种3.5×106个MDA-MB-436细胞,建立MDA-MB-436-异种移植物肿瘤。当肿瘤达到约100mm3时,将小鼠随机分为五个治疗组(每组8只动物):1)口服载体对照;2) 口服10mg/kg PO BID AMXI-5001,5天on/2天OFF周期;3) 口服50mg/kg PO BID AMXI-5001,5天on/2天OFF周期;4) 口服奥拉帕尼50mg/kg BID,5天on/2天OFF周期;5) 每周一次(Q1W)腹腔注射1 mg/kg长春花碱。所有动物均在31天的给药期内接受治疗。每只动物的肿瘤大小每周测量两次(图4A)。 AMXI-5001表现出显著的、剂量依赖性的肿瘤生长抑制作用。除了肿瘤生长抑制外,AMXI-5001在该模型中还表现出明显的肿瘤消退。对于50 mg/kg AMXI-5001组,所有肿瘤均完全消退。到给药计划的第31天,肿瘤要么太小而无法准确测量,要么无法触及。与奥拉帕尼相比,AMXI-5001在10或50 mg/kg/剂量BID下观察到的抗肿瘤生长效果优于奥拉帕尼在50 mg/kg/剂BID下的效果。与AMXI-500l相比,奥拉帕尼在此模型中没有表现出任何肿瘤消退活性,而以1 mg/kg Q1W剂量给药的长春碱在此模型中对肿瘤生长没有显著影响(图4A)。 以10和50mg/kg/剂BID的AMXI-5001剂量在5天on/2天OFF周期内治疗31天的动物,没有表现出任何与化合物相关的毒性的身体症状,奥拉帕尼和长春花碱治疗的动物也没有。在整个治疗过程中,任何一组的体重都没有显著影响(图4B)。 为了评估治疗相关的组织学变化,在治疗结束时采集的肿瘤石蜡切片用苏木精-伊红(H&E)染色。H&E染色显示,用50 mg/kg剂量的AMXI-5001治疗的肿瘤出现了严重的出血性肿瘤坏死(图4C)。在治疗结束时,用50mg/kg的AMXI-5001治疗的小鼠收获的肿瘤是不可触及的。该组的肿瘤切片显示,少数剩余的异种移植物癌症细胞通过广泛的纤维化分散,周围是宿主动物的皮肤、骨骼肌和脂肪。在更高的放大倍数下,这些肿瘤的照片显示了显著的治疗效果,有坏死和出血的证据,有含铁血黄素填充的巨噬细胞积聚、胆固醇肉芽肿、慢性炎症和纤维化(图4C)。用10mg/kg的AMXI-5001治疗的小鼠的肿瘤也显示出异种移植物细胞密度降低,并出现坏死、炎症和纤维化区域。来自载体治疗、奥拉帕尼治疗或长春碱治疗的小鼠的肿瘤切片主要含有活的肿瘤细胞,没有任何纤维化(图4C)。 在研究结束时,切除肿瘤并进行处理,以分析微管丝的形成。石蜡包埋的肿瘤切片用α/β微管蛋白特异性抗体染色,并通过荧光显微镜观察。细胞核用4',6-二脒基-2-苯基吲哚(DAPI)染色。以10mg/kg或50mg/kg BID重复口服AMXI-5001剂量,对MDA-MB-436细胞衍生肿瘤中的微管丝形成产生了显著的剂量依赖性抑制作用(图S34)。长春花碱治疗还诱导MDA-MB-436肿瘤中微管丝的显著抑制。相反,以50mg/kg BID剂量重复口服奥拉帕尼对肿瘤细胞微管丝的形成没有影响。 AMXI-5001治疗的药效学参数也使用本研究中每组异种移植物肿瘤标本制备的裂解物进行测定。对于PARP抑制,评估标准杆数水平,对于微管不稳定,通过蛋白质印迹分析评估总α/β微管蛋白表达水平及其特异性对应抗体。10或50 mg/kg的AMXI-5001治疗以剂量依赖性方式显著抑制了MDA-MB-436衍生肿瘤中标准杆数的表达(图S35)。AMXI-5001治疗还导致MDA-MB-436衍生肿瘤中总α/β微管蛋白表达水平显著降低,且呈剂量依赖性(图S35)。奥拉帕尼治疗导致标准杆数表达的显著抑制,但对MDA-MB-436来源的肿瘤中的微管蛋白表达没有影响。相反,长春碱治疗对MDA-MB-436衍生肿瘤中标准杆数水平或总微管蛋白表达没有影响(图S35)。与PARP抑制作用一致,与赋形剂对照治疗相比,AMXI-5001治疗和奥拉帕尼治疗均显著抑制了细胞周期检查点蛋白(CHFR)的肿瘤蛋白表达(图S35)。 总的来说,上述结果表明,AMXI-5001以剂量依赖的方式调节其在肿瘤体内的预期靶点。这些结果还表明,AMXI-5001的抗肿瘤活性可能归因于其通过同步抑制肿瘤细胞中的PARP和微管聚合的双重作用机制。 单药AMXI-5001治疗MDA-MB-436异种移植物中已建立的大肿瘤(600-1300mm3)完全消退[1] 基于上述研究中MDA-MB-436异种移植物在开始给药前肿瘤分期为100-150mm3时的抗肿瘤疗效,在更大、更成熟的肿瘤中测试了AMXI-5001的抗肿瘤作用(图5A)。 在上述研究的31天给药期结束时,来自肿瘤大小为554-1318mm3的对照组的8只小鼠中有5只在5天on/2天OFF周期内以50mg/kg/剂量BID的剂量接受AMXI-5001,来自长春碱组的2只小鼠和来自对照组的1只小鼠(肿瘤大小458-953mm3)按相同的时间表接受赋形剂对照。值得注意的是,从治疗开始的第一周开始,所有用AMXI-5001治疗的大肿瘤都表现出逐渐和近似完全的肿瘤消退(图5A)。值得注意的是,在AMXI-5001治疗后,小鼠停止治疗2周后没有肿瘤复发的迹象(图5A)。相反,在新的载体治疗组中,肿瘤继续快速生长,在载体治疗开始后不到三周,由于肿瘤过度生长和随之而来的动物健康状况不佳,小鼠被终止。与赋形剂对照治疗组相比,AMXI-5001以50mg/kg/剂量BID在整个治疗过程中对体重没有显著影响(图5B)。 在MDA-MB-436异种移植物模型中,AMXI-5001的抗肿瘤效果优于单药PARP和微管抑制剂的联合治疗[1] 本研究旨在比较AMXI-5001治疗与单剂奥拉帕尼(一种临床PARP抑制剂)或单剂紫杉醇(一种强效临床微管聚合抑制剂)的体内抗肿瘤疗效,以及奥拉帕尼和紫杉醇在MDA-MB-436异种移植物模型中的联合治疗。奥拉帕尼和紫杉醇以临床相关剂量和时间表给药。 AMXI-5001在所有接受治疗的动物中诱导了完全或接近完全的肿瘤消退,这种效果优于单药奥拉帕尼或紫杉醇,或两种药物的联合治疗(图6A)。在这种肿瘤模型中,单药紫杉醇治疗显示出较弱的抗肿瘤活性,而单药奥拉帕尼治疗诱导了明显但适度的肿瘤生长抑制。奥拉帕尼和紫杉醇的联合治疗比单独使用这两种药物更能抑制肿瘤生长。然而,奥拉帕尼和紫杉醇的联合治疗未能诱导肿瘤消退,肿瘤继续生长,尽管其生长速度比载体治疗或单一药物(奥拉帕尼或紫杉醇)治疗的肿瘤慢。AMXI-5001在该模型中比单药奥拉帕尼与单药紫杉醇的组合更有效。 用AMXI-5001、单药奥拉帕尼或奥拉帕尼与紫杉醇联合治疗的动物没有显示出任何明显的治疗相关毒性(图6B)。然而,单药紫杉醇组8只小鼠中有3只在首次静脉注射紫杉醇(30mg/kg)后几分钟内出现嗜睡、对外部刺激缺乏反应、意识丧失和呼吸困难。该组3只受影响的动物被安乐死。然而,在单药紫杉醇治疗组中,其余5只动物在治疗期间均未表现出任何明显的治疗相关毒性。与赋形剂对照治疗相比,在整个治疗过程中,任何治疗方式对体重都没有明显的治疗相关影响。 |
| 酶活实验 |
体外激酶抑制试验[1]
AMXI-5001抑制脱靶激酶的潜力在156项重组人激酶活性和结合试验中进行了评估,包括细胞质和受体酪氨酸激酶、丝氨酸/苏氨酸激酶和脂质激酶。激酶谱分析使用Life Technologies的SelectScreen®谱分析服务进行,广泛覆盖人类激酶。激酶活性测定测量肽磷酸化或ADP产生,而激酶结合测定监测ATP位点结合探针的位移。活性测定中使用的ATP浓度在每种激酶的实验确定的表观米氏常数(K m app)值的2倍以内,而结合测定中使用了竞争性结合示踪剂浓度在实验确定的解离常数(K d)值的3倍以内。AMXI-5001在8µM下对每种激酶进行了三次测试,并报告了平均%抑制值。对于选定的激酶,使用与单点试验中使用的激酶测定相同的激酶测定法进行10点抑制剂滴定,以确定提供50%抑制的抑制剂浓度(IC50)。 PARP2抑制试验[1] 使用市售的微孔板测定试剂盒并按照制造商提供的说明测定测试化合物对PARP2的抑制作用。随后,使用GraphPad Prism进行非线性拟合后,确定了PARP2抑制的IC50。 PARP酶测定[1] 使用市售的微孔板测定试剂盒并按照制造商提供的说明测定测试化合物对PARP1的抑制作用。简而言之,各种测试化合物的储备溶液是在二甲基亚砜(DMSO)中制备的。对于该测定,每个条孔填充10µL抑制剂溶液、20μL稀释的PARP1酶(提供0.5单位/孔)和25µL PARP鸡尾酒(由生物素化的NAD、pH 8.0的Tris-Cl中的活化DNA和EDTA组成)。将条孔在室温下孵育60分钟,然后用磷酸缓冲盐水(PBS:Na2HPO4、NaH2PO4和NaCl)和0.1%Triton X-100洗涤4次,用PBS洗涤2次。然后,向每个孔中加入50µL稀释的Strep-HRP(阻断溶液),并在室温下进一步孵育条带60分钟。用PBS、0.1%Triton X-100和PBS洗涤孔各2次后,将其与50µL TACS Sapphire™比色底物混合,并在黑暗中静置15分钟。通过向每个孔中加入50μL 0.2 N盐酸(HCl)停止反应后在450nm处测量吸光度。通过用等体积的DMSO代替测试溶液进行平行实验,以验证载体对酶活性的影响。所有检测均在至少两个单独的场合进行,每次重复。随后,使用GraphPad Prism进行非线性拟合后,确定PARP1抑制的IC50。这些研究的结果以μM为单位报告。DMSO用作阴性对照。临床批准的PAPR抑制剂(olaparib、talazoparib、niraparib或rucaparib)用作PARP抑制的阳性对照。在某些情况下,紫杉醇也被用作阴性对照。 体外微管蛋白聚合试验[1] 根据制造商的方案,使用基于体外荧光的微管蛋白聚合测定试剂盒来监测微管蛋白向微管的时间依赖性聚合。在存在或不存在测试化合物的情况下,制备含有猪脑微管蛋白、荧光报告蛋白和GTP的反应混合物。在微管蛋白聚合后,监测荧光增强,这是由于在聚合过程中将荧光报告子掺入微管中造成的。在Biotek Synergy平板读数器中以1分钟的间隔测量荧光发射(在360nm波长下激发和在460nm波长下消除)1小时45分钟。在上述条件下,约45%的微管蛋白聚合,为检测聚合增强剂和抑制剂留下了灵活性。DMSO用作阴性对照。紫杉醇被用作微管蛋白聚合增强的阳性对照。长春花碱被用作微管蛋白聚合抑制的阳性对照。测量了聚合过程中荧光报告子掺入微管的情况。使用GraphPad Prism 6程序绘制360nm激发和460nm消除波长下的荧光吸光度与反应时间的关系图。使用GraphPad Prism进行非线性拟合后确定Vmax。DMSO对照的Vmax设定为100%聚合。将化合物Vmax相对于DMSO对照Vmax的百分比与化合物浓度作图。随后,使用GraphPad Prism进行非线性拟合后,确定了微管蛋白聚合抑制剂的IC50。 竞争性MS结合分析[1] 为了确定AMXI-5001在微管蛋白上的结合位点,如前所述进行了竞争性MS结合分析,但秋水仙碱、长春花碱和紫杉醇的结合条件略有不同。该测定用于评估秋水仙素、长春花碱和紫杉醇的微管蛋白结合,并鉴定AMXI-5001结合这三个结合位点中的哪一个。该方法涉及一个非常简单的步骤,即使用超滤将未结合的配体与大分子分离。使用高灵敏度和特异性的HPLC-MS/MS方法可以准确地测定流经级分中的未结合配体。简言之,将诺考达唑-秋水仙素和长春花碱与微管蛋白在37°C的孵育缓冲液中孵育1小时。将紫杉醇与已完成的微管蛋白在培养缓冲液中于37°C孵育1小时时。通过将微管蛋白与GTP在培养缓冲区中于37℃孵育1个小时来制备已完成的小管蛋白。使用不同浓度的诺考达唑、长春新碱和多西他赛分别与秋水仙素、长春花碱和紫杉醇的微管蛋白结合竞争。孵育后,将反应样品在Ultracel-30微浓缩器中离心。如下所述,收集流动(未结合的配体)并通过HPLC-MS/MS进行分析。检测了不同浓度的AMXI-5001,以分别与秋水仙碱、长春花碱和紫杉醇微管蛋白结合竞争。在没有任何竞争对手的情况下,竞争对手或AMXI-5001抑制配体结合的能力表示为未结合配体对照的百分比。 |
| 细胞实验 |
细胞标准杆数蛋白质印迹分析[1]
AMXI-5001对细胞标准杆数水平的影响也通过标准蛋白质印迹程序进行评估。简言之,将癌症细胞在6孔板中培养过夜,然后用载体对照或测试化合物孵育24小时。然后洗涤细胞,制备细胞裂解物用于蛋白质印迹,如下所述。使用BCA法定量每种细胞裂解物中的蛋白质浓度。随后使用标准程序进行免疫印迹。通过SDS-PAGE分解总共10μg的蛋白质,将其转移到聚偏二氟乙烯(PVDF)膜上,并用抗-PAR和第二抗体进行探测。临床批准的PARPi(olaparib、talazoparib或rucaparib)用作标准杆数抑制和γH2AX诱导的阳性对照。β-actin作为负载对照。 细胞PARP-DNA捕获试验[1] 为了评估AMXI-5001的PARP捕获能力,如前所述进行了标准细胞捕获试验。简言之,将癌症细胞在6孔板中培养过夜,然后用烷基化剂甲基甲磺酸酯(MMS)和载体对照共同处理,或用烷基化试剂和不同浓度的测试PARPi共同处理1或3小时。然后洗涤细胞并通过胰蛋白酶消化收集细胞。随后,按照制造商的方案,使用Thermo Scientific亚细胞蛋白分级试剂盒制备染色质组分。对样品进行蛋白质浓度标准化,并通过抗PARP1、抗TOP1和抗H3的免疫印迹进行分析。临床批准的PARPi(olaparib、talazoparib或rucaparib)用作PARP-DNA捕获的阳性对照。为了量化PARPi诱导的人癌症细胞中PARP1-DNA捕获,对免疫印迹进行密度测定。将DNA结合的PARP1水平标准化为总细胞PARP1水平(染色质结合的PARP1+未结合的PARP 1)。每个实验至少进行三次独立的时间。代表性结果见下文结果部分。 细胞滴度Glo测定[1] AMXI-5001的抗增殖活性在一组110个不同来源的癌症细胞系中进行评估,这些细胞系对于BRCA-1或BRCA-2表达或表达这两个基因的突变形式是熟练的或有缺陷的。在细胞暴露于8 nM至5μM的AMXI-5001剂量3天或6天后评估增殖。简而言之,在加入AMXI-5001的系列稀释液之前,将5000或1000个细胞(分别在3天或6天的暴露中)在96孔板中培养,并在37°C和5%CO2下孵育。然后将细胞在37°C和5%CO2下孵育3-6天。通过Cell Titer-Glo试验评估细胞存活率。通过读取GloMax Luminametor上的平板来确定活细胞的数量。细胞生长以相对于载体(DMSO对照)处理的细胞的百分比生长表示。使用GraphPad Prism进行非线性拟合后,确定抑制细胞生长50%所需的浓度(IC50)。DMSO处理的细胞用作载体对照。MTA(紫杉醇和长春花碱)以及临床PARP(奥拉帕尼、塔拉佐帕尼、尼拉普里布和鲁卡帕里布)用作对照。 克隆形成试验[1] AMXI-5001对细胞活力的影响也使用卵巢、非小细胞肺癌和前列腺来源的细胞系中的集落形成测定进行评估。简而言之,细胞系在37°C下在0.1%(v/v)DMSO(载体对照)或0.008、0.04、0.2、1、5 mM AMXI-5001中孵育,直至形成>50个菌落(6-28天)。通过初步实验确定接种细胞密度,并将其定义为在0.1%(v/v)DMSO(载体对照)中培养细胞后产生线性细胞生长的接种密度。通过使用ImageJ软件计数结晶紫染色的集落,使用集落形成测定法测定细胞存活率。IC50值定义为与载体对照细胞(100%细胞存活率)相比,产生50%细胞存活率的AMXI-5001浓度。临床PARP(Olaparib、Talazoparib、Niraparib和Rucaparib)用作对照。 伤口愈合试验/划痕试验[1] AMXI-5001抑制细胞迁移的能力使用体外划痕试验来测试,该试验通过恢复生长成单层的A549肺癌细胞中的划痕伤口来评估细胞迁移。简言之,将A549肺癌细胞接种到12孔板上。当细胞融合达到约80%及以上时,使用200μl移液管尖端在每个培养板上刮擦细胞层,形成划痕。受伤后,用PBS清洗细胞以去除碎片。受伤的培养物在37°C下单独在培养基(未处理的对照)或含有0.1%(v/v)DMSO(DMSO对照)或0.04、0.2、1 mM AMXI-5001的培养基中孵育24小时。随后,从每个划痕中随机选取3个区域(40Å~),并通过显微镜观察以评估细胞迁移能力。临床PARP(奥拉帕尼和塔拉佐帕尼)和临床MTA(紫杉醇和长春花碱)用作细胞迁移抑制的阳性对照。 细胞周期分析[1] 评估了AMXI-5001对MDA-MB-436、OVCAR8和A549细胞分裂过程中细胞周期进程的影响。简而言之,细胞在T75培养瓶中生长,用0.1%DMSO(载体对照)、长春花碱或AMXI-5001以不同浓度处理24小时。孵育后,将细胞胰蛋白酶化,用PBS洗涤并固定。固定后,将细胞离心,用PBS洗涤一次,并用碘化丙啶染色。最后,使用流式细胞术分析处理细胞在细胞周期不同阶段(G1、S、G2/M)的分布。测定AMXI-5001治疗24小时前后每个有丝分裂期的细胞百分比。MTA(紫杉醇和长春碱)用作细胞周期阻滞的阳性对照。 细胞检查点和信号蛋白的蛋白质印迹分析[1] 为了更好地理解AMXI-5001调节癌症细胞中细胞周期阻滞的机制,对细胞裂解物进行了标准西方分析,以评估各种检查点相关蛋白和细胞信号蛋白的状态(图3)。简言之,癌症细胞在无血清培养基中的6孔板中培养过夜,然后与载体对照或测试化合物孵育24小时。然后洗涤细胞,制备细胞裂解物用于蛋白质印迹,如下所述。使用BCA法定量每种细胞裂解物中的蛋白质浓度。随后使用标准程序进行免疫印迹。SDS-PAGE共分离10μg蛋白质,转移到聚偏二氟乙烯(PVDF)膜上,用检查点蛋白对应抗体和二抗进行检测。临床批准的PARPi(奥拉帕尼、塔拉佐帕尼)或MTA(紫杉醇、长春碱)用作对照。β-actin和GAPDH用作负载对照。 流式细胞术分析细胞表面标记染色[1] A549细胞在100mm培养皿中培养过夜,然后与载体对照或AMXI-5001以1mM或5mM孵育24小时或48小时,然后通过胰蛋白酶消化收获,如下文方法部分所述。对于PDL1和死亡受体(DR4和DR5)细胞表面染色,用PBS洗涤收获的细胞,用细胞染色缓冲液中的相应抗体染色,并进行流式细胞术分析。BD CellQuest Pro软件用于流式细胞术数据分析。 |
| 动物实验 |
Pharmacokinetic evaluations of AMXI-5001 in mice[1]
The objective of these studies was to determine pharmacokinetic and bioavailability profile following a single dose oral administration of either free base form or HCl salt forms of AMXI-500 in BALB/c mice. The In-Life procedures for these studies were conducted using Explora Bioloabs contract research services. The in vitro bioanalyses of the plasma samples were performed using Integrated Analytical Solutions contract research services. Briefly, AMXI-5001 free base and HCl salt form were formulated either as NMP/CMC suspension or in 10% TPGS suspension, respectively and administered orally to female BALB/c mice. The formulations protocols used for the PK studies are described below. The bioavailability for AMXI-5001 free base form was assessed following a single oral dose administration at either 50 or 100 mg/kg per mouse. The bioavailability for AMXI-5001 HCL form was assessed following a single oral dose administration at 50 mg/kg per mouse. Blood samples were collected at pre-dose, 0.5, 1, 2, 4, 8 and 24 hours after AMXI-5001 single dose oral administration. For each mouse, one-two time points were assigned. Each group had 3 blood samples per time point. The first blood collection was survival bleed and the second blood collection was terminal. Blood samples were collected into tubes containing K2EDTA anticoagulant and stored on wet ice until centrifuged and processed for plasma. Plasmas were stored at -80°C until analysis. The peak concentration (Cmax), the time to maximum concentration (Tmax), the half-life, and the AUC were determined from composite mean plasma concentration-time data. All doses and plasma concentrations of AMXI-5001 were presented as free base. Pharmacodynamic evaluations of AMXI-5001 in mice bearing MDA-MB-436 tumors[1] The primary objective of this study was to determine the relationship between AMXI-5001 dose, plasma concentration, tumor concentration and inhibition of PARP and microtubule polymerization In vivo in MDA-MB-436 Xenograft Tumors. The In-Life procedures for this study were conducted using Explora Bioloabs contract research services. The in vitro bioanalyses of the plasma samples were performed using Integrated Analytical Solutions contract research services. Briefly, AMXI-5001 HCL salt form was formulated in 10% TPGS suspension, as described below and administered orally to female athymic nude mice bearing established MDA-MB-436 Xenograft Tumors (When tumors reach average volume of 500 mm3). AMXI-5001 suspension was administered orally BID at 6.25, or 12.5, or 25 mg/kg per mouse for 5 days. At 3 hr post the 4th and 10th dose, n=3 mice per group, or each AMXI-5001-dose and the vehicle control group, were euthanized and blood collected in EDTA tubes for plasma processing. At necropsy the tumor tissues from all groups, as well as stomach, small intestine, large intestine, caecum, kidneys, and liver from the vehicle control group and the group treated with AMXI-5001 at 50 mg/kg BID, were individually collected, placed into cryotubes and flash frozen in liquid nitrogen. The frozen plasma and tissue samples were kept at -80°C until bioanalysis for AMXI-5001 concentration. Plasma samples were analyzed by high performance liquid chromatography (HPLLC) in conjunction with a triple quadrupole mass spectrometer that used electrospray ionization in tandem with positive ionization (MS/MS). The lower limit of quantitation (LLOQ) was 5 ng/ml. The quantitative range of the assay was 5 to 10000 ng/ml. In vivo xenograft models[1] In the present studies, the MDA-MB-436 (triple negative human breast carcinoma with BRCA1 mutation) was utilized to explore the efficacy and potency of AMXI-5001 with regard to tumor growth inhibition and regression in vivo. The In-Life procedures for these studies were conducted using the Explora Biolabs contract research services. The standard experimental design for these studies involved twice daily (BID) daily oral administration (PO) of AMXI-5001, following a 5-day ON and 2-day OFF cycles, beginning when the established solid tumors were staged (~100 -150 mm3 for most xenografts, ~700 mm3 and up to 1500 mm3 for xenograft regression studies). Throughout the dosing period of 10-60 days, tumor size and body weight were measured twice weekly. At the end of the study, tumors were resected and processed for analysis of microtubule filament formation. Sections from paraffin embedded tumors were stained with an antibody specific to for alpha/beta-tubulin, and visualized by fluorescent microscopy. In order to assess treatment related histological changes, paraffin sections of harvested tumors taken at the end of treatment were also stained with hematoxylin-eosin (H&E) and analyzed by a certified pathologist. Pharmacodynamic parameters for AMXI-5001 treatment, were also determined using lysate prepared with xenograft tumor specimens from each group in this study. For poly {adenosine diphosphate (ADP)}-ribose polymerase (PARP) inhibition (PARPi), Poly (ADP-ribose) (PAR) levels were evaluated, and for microtubule destabilization, total alpha/beta tubulin expression levels were assessed by Western blot analyses with their specific corresponding antibodies. HT PARP in vivo pharmacodynamic assay[1] The HT PARP In Vivo Pharmacodynamic Assay is a high-throughput, chemiluminescent ELISA designed to quantify poly (ADP-ribose) (PAR) in cellular extracts. The assay employs a two-site sandwich technique in which two different anti-PAR antibodies are used to capture and detect the target analyte. This assay is useful for measuring PAR in extracts from peripheral blood mononuclear cells (PBMC), cultured cells, and tissues. Additionally, this assay can be used to monitor the efficacy of PARPi or anti-cancer drugs on cellular PAR formation and cancer cell cytotoxicity. The inhibitory effect of AMXI-5001 on cellular PAR formation was quantified with this HT PARP In Vivo Pharmacodynamic Assay in accordance with the instructions provided by the manufacturer. Each ELISA plate contains serial dilutions of purified PAR standard used to plot the PAR standard curve. The net mean RLU (Relative Light Units) values of the PAR standards were calculated by subtracting the background (without PAR) from the RLU values, then plotted as a function of the corresponding PAR values (pg/ml). The PAR standard curve was plotted using the GraphPad Prism 6 program. Typically, the linear dynamic range for the PAR standard curve is from 10 to 1000 pg/ml. The net RLU values for each cell lysate sample was calculated by subtracting the background from the RLU values. Subsequently, the PAR levels in each sample was determined using the PAR standard curve. The PAR level of DMSO-treated control was set to 100% PAR level. The percentages of test compounds-treated samples over DMSO-treated control PAR levels were plotted against the compound concentrations. Subsequently, the IC50s for the cellular PAR formation inhibitors were determined after non-linear fit using GraphPad Prism. |
| 药代性质 (ADME/PK) |
Metabolism and PK properties of AMXI-5001 [1]
Currently, there are no approved orally bioavailable microtubule targeting agent. One of the objectives of our dual PARP and microtubule inhibitor discovery program was to develop an orally bioavailable dual PARP and microtubule inhibitor and to improve metabolic stability, PK properties and oral bioavailability over existing PARP1/2 inhibitors. In vitro metabolism studies of AMXI-5001 in hepatocytes from rats, dogs, cynomolgus monkeys, and humans demonstrated that AMXI-5001 had excellent liver stability; The half-life of AMXI-5001 (2 μM) during incubation with hepatocytes (500,000 cells/mL) from Sprague-Dawley rats, beagle dogs, cynomolgus monkeys, or humans was estimated to be 217 min, 812 min, 185 min, and 417 min, respectively. After incubation 1 μM concentration for 120 min at 37°C, the percentage of AMXI-5001 remaining was 63.6%, 85.4%, 64.1%, and 78.5% for rat, dog, monkey, and human hepatocytes, respectively (Not shown). These data suggest AMXI-5001 will have human clearance approximately similar to that of the animal species. AMXI-5001 has been given orally to rats and dogs at various dose levels (manuscript in preparation). AMXI-5001 was absorbed and bioavailable in all species tested. In rats and dogs, exposure increased with the increase in dose level, supporting the use of these species in the toxicity studies of AMXI-5001. A variety of formulations were tested, eventually leading to selection of 10% TPGS (D-α-tocopherol polyethylene glycol-1000-succinate; Vitamin E) in 0.01 N HCl, pH 2.1-2.3, because of its suitable toxicity profile and ability to deliver adequate AMXI-5001 systemic exposure. AMXI-5001 demonstrated an absolute bioavailability in rats and dogs of 31% and 64%, respectively with this formulation, and PK properties that would predict a human half-life that is sufficient to support a regimen of twice daily administration (manuscript in preparation). In vitro studies assessing the potential for inhibition of human cytochrome P450 enzymes (CYP450s) showed that AMXI-5001 did not inhibit any of the major human hepatic CYP450 enzymes CYP1A2, CYP2B6, CYP2D6, and CYP3A4/5. There was weak (and not time-dependent) inhibition of CYP2C9, and CYP2C19 (Data not shown). Overall, AMXI-5001 demonstrated excellent metabolic stability, oral bioavailability and PK properties. AMXI-5001 pharmacokinetic assessment [1] To support the in vivo primary pharmacodynamic studies in xenograft models, pharmacokinetic parameters of AMXI-5001 were determined in female BALB/C mice. AMXI-5001 free base or hydrochloride salt forms were formulated either as N-methyl-pyrrolidine/carboxymethylcellulose (NMP/CMC) suspension or in 10% D-α-tocopherol polyethylene glycol-1000-succinate; Vitamin E (TPGS) suspension, respectively and administered orally. AMXI-5001 was absorbed and bioavailable in this strain of mice, enabling its testing in murine xenograft models with oral dosing (Table S18). |
| 参考文献 | |
| 其他信息 |
Poly (ADP-ribose) polymerase (PARP) has recently emerged as a central mediator in cancer resistance against numerous anticancer agents to include chemotherapeutic agents such as microtubule targeting agents and DNA damaging agents. Here, we describe AMXI-5001, a novel, highly potent dual PARP1/2 and microtubule polymerization inhibitor with favorable metabolic stability, oral bioavailability, and pharmacokinetic properties. The potency and selectivity of AMXI-5001 were determined by biochemical assays. Anticancer activity either as a single-agent or in combination with other antitumor agents was evaluated in vitro. In vivo antitumor activity as a single-agent was assessed in a triple-negative breast cancer (TNBC) model. AMXI-5001 demonstrates comparable IC50 inhibition against PARP and microtubule polymerization as clinical PARP inhibitors (Olaparib, Rucaparib, Niraparib, and Talazoparib) and the potent polymerization inhibitor (Vinblastine), respectively. In vitro, AMXI-5001 exhibited selective antitumor cytotoxicity across a wide variety of human cancer cells with much lower IC50s than existing clinical PARP1/2 inhibitors. AMXI-5001 is highly active in both BRCA mutated and wild type cancers. AMXI-5001 is orally bioavailable. AMXI-5001 elicited a remarkable In vivo preclinical anti-tumor activity in a BRCA mutated TNBC model. Oral administration of AMXI-5001 induced complete regression of established tumors, including exceedingly large tumors. AMXI-5001 resulted in superior anti-tumor effects compared to either single agent (PARP or microtubule) inhibitor or combination with both agents. AMXI-5001 will enter clinical trial testing soon and represents a promising, novel first in class dual PARP1/2 and microtubule polymerization inhibitor that delivers continuous and synchronous one-two punch cancer therapy with one molecule.[1]
AMXI-5001 demonstrated a remarkable In vivo preclinical anti-tumor activity in BRCA mutated triple negative breast cancer (TNBC) model, a cancer with currently no effective therapy. Oral administration of single agent AMXI-5001 induced complete regression of established tumors, including exceedingly large tumors (Figures 4, 5 and 6). Most importantly, none of the AMXI-5001 treated mice had tumor re-growth until the end of the study, two weeks after AMXI-5001 dosing stopped. Furthermore, AMXI-5001 resulted in superior anti-tumor effects when compared to either single agent (PARP inhibitor (Olaparib) or microtubule targeting agent (Paclitaxel or Vinblastine)) or combination treatment with both agents which were given at clinically relevant doses (Figures 4A, 6A). Importantly, the AMXI-5001 anti-tumor effect was achieved with tolerable toxicity, evidence of PARP and microtubule inhibition in vivo in tumors and favorable PK properties that allow twice-a-day oral dosing in human patients. AMXI-5001 is the first in class small-molecule inhibitor reported to date that offers a continuous and synchronous PARP and microtubule polymerization inhibitions, and thus results in synthetic lethality, particularly in cancer cells vulnerable to DNA damage. The discovery and characterization of AMXI-5001 as an orally bioavailable dual PARP and microtubule polymerization inhibitor, provides a welcome addition to the oncology field and we believe the pharmacological properties of AMXI-5001 warrant further investigation, and its advancement into clinical studies in cancer patients. [1] |
| 分子式 |
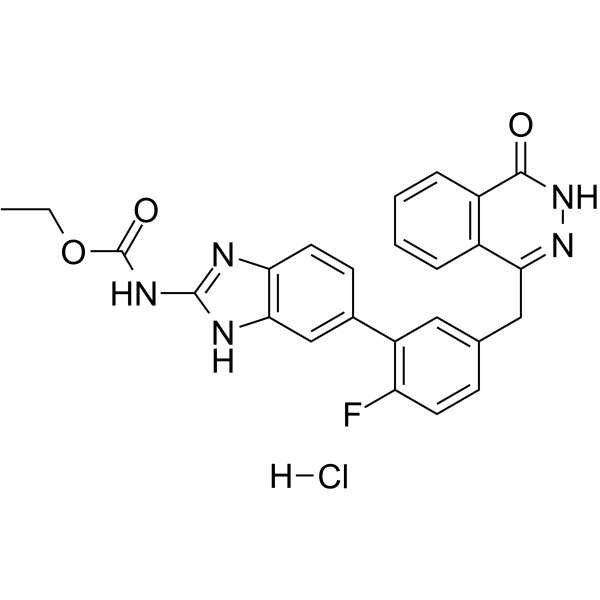
C25H21CLFN5O3
|
|---|---|
| 分子量 |
493.92
|
| 精确质量 |
493.13169
|
| PubChem CID |
163196447
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
108 Ų
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
35
|
| 分子复杂度/Complexity |
795
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
DCTVJMPYKWHEFD-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C25H20FN5O3.ClH/c1-2-34-25(33)29-24-27-20-10-8-15(13-22(20)28-24)18-11-14(7-9-19(18)26)12-21-16-5-3-4-6-17(16)23(32)31-30-21;/h3-11,13H,2,12H2,1H3,(H,31,32)(H2,27,28,29,33);1H
|
| 化学名 |
ethyl N-[6-[2-fluoro-5-[(4-oxo-3H-phthalazin-1-yl)methyl]phenyl]-1H-benzimidazol-2-yl]carbamate;hydrochloride
|
| 别名 |
AMXI-5001 (hydrochloride); CHEMBL5184668; AMXI5001 hydrochloride;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0246 mL | 10.1231 mL | 20.2462 mL | |
| 5 mM | 0.4049 mL | 2.0246 mL | 4.0492 mL | |
| 10 mM | 0.2025 mL | 1.0123 mL | 2.0246 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























