| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 体外研究 (In Vitro) |
在 CHO 细胞中,溴隐亭增加 D2 多巴胺受体的表达,然后与 [35S]-GTPγS 结合,pEC50 为 8.15±0.05[1]。另一种有效的脑一氧化氮合酶抑制剂是溴隐亭。结果表明,麦角生物碱溴隐亭(BKT)对诱导型巨噬细胞NOS作用较弱(IC50>100 μM),但它是纯神经元一氧化氮合酶(NOS)的有效抑制剂(IC50=10±2 μM)[2 ]。已发现至少一种人类细胞色素P450酶被溴隐亭抑制。强效 CYP3A4 抑制剂溴隐亭的相互作用 IC50 值为 1.69 μM [3]。
|
|---|---|
| 体内研究 (In Vivo) |
与对照组相比,腹腔注射2mg/kg多巴胺激动剂溴隐亭的组表现出显着的抗不动效果。当 7 天 MPE 治疗最后一次给药后 30 分钟给予溴隐亭并进行 FST 时,这种多巴胺能激动剂对 MPE(200 mg/kg,口服)的抗不动作用显示出显着且剂量-与单独 MPE 治疗相比,依赖性增强。与对照组相比,腹膜内 (ip) 2 mg/kg 多巴胺激动剂溴隐亭治疗组的不动时间显着缩短。当用 MPE 预处理 7 天后给予溴隐亭(100 和 200 mg/kg,po)时,与单独 MPE 治疗相比,MPE 的抗不动效果显着且呈剂量依赖性增强 [4]。当腹腔注射溴隐亭时,与假手术组相比,CCI-IoN 组的疼痛评分出现显着的剂量依赖性下降(0.1 mg 和 1 mg/Kg)。这种效果持续了六个小时。当应用最高剂量时,分数下降最多(P<0.01)。 DR1激动剂SKF8129用作阳性对照。腹膜内给药与假手术(盐水注射)相比,SMA 评分没有显着上升。当宫颈内给予溴隐亭时,SMA 评分比使用盐水注射作为假手术时低得多。溴隐亭的半衰期为 20 分钟。腹腔注射溴隐亭后,CCI-IoN α + α 6-OHDA 损伤组的 SMA 评分较假手术组显着下降,且呈剂量依赖性。撞击期间已经过去了六个小时。 SKF81297 的给药导致更高的异常性疼痛评分。与假手术(给大鼠注射生理盐水)相比,脑池内输注溴隐亭可显着降低 SMA 评分,且其效果持续 30 分钟[5]。
|
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服剂量约有28%被吸收;然而,由于显著的首过效应,仅有6%的口服剂量以原形进入体循环。溴隐亭及其代谢物在口服后10分钟即可出现在血液中,并在1-1.5小时内达到血浆峰浓度。口服后2小时内血清催乳素水平可能下降,并在8小时后达到最大效应。肢端肥大症患者单次口服 2.5 mg 后,生长激素浓度在 1-2 小时内降低,且降低的生长激素浓度至少持续 4-5 小时。 原药及其代谢物几乎完全经肝脏排泄,仅有 6% 经肾脏排泄。 代谢/代谢物 在肝脏中完全代谢,主要通过酰胺键水解生成麦角酸和肽片段,两者均无活性且无毒。溴隐亭经细胞色素P450 3A4代谢,主要通过胆汁分泌经粪便排出。 溴隐亭已知的人体代谢物包括5-溴-N-[2,10-二羟基-7-(2-甲基丙基)-5,8-二氧代-4-丙-2-基-3-氧杂-6,9-二氮杂三环[7.3.0.02,6]十二烷-4-基]-7-甲基-6,6a,8,9-四氢-4H-吲哚并[4,3-fg]喹啉-9-甲酰胺和5-溴-N-[2,11-二羟基-7-(2-甲基丙基)-5,8-二氧代-4-丙-2-基-3-氧杂-6,9-二氮杂三环[7.3.0.02,6]十二烷-4-基]-7-甲基-6,6a,8,9-四氢-4H-吲哚并[4,3-fg]喹啉-9-甲酰胺。 溴隐亭在肝脏中完全代谢,主要通过酰胺键水解生成麦角酸和肽片段,两者均无活性且无毒。溴隐亭经细胞色素P450 3A4代谢,主要通过胆汁分泌经粪便排泄。 排泄途径:原药及其代谢物几乎完全经肝脏排泄,仅有 6% 经肾脏排泄。 半衰期:2-8 小时 生物半衰期 2-8 小时 |
| 毒性/毒理 (Toxicokinetics/TK) |
毒性概述
多巴胺D2受体是一种7次跨膜G蛋白偶联受体,与Gi蛋白相关。在泌乳细胞中,多巴胺D2受体的激活会导致腺苷酸环化酶的抑制,从而降低细胞内cAMP浓度并阻断IP3依赖的Ca2+从细胞内储存的释放。细胞内钙水平的降低也可能是通过抑制电压门控钙通道的钙内流而非腺苷酸环化酶的抑制来实现的。此外,受体激活会阻断p42/p44 MAPK的磷酸化并降低MAPK/ERK激酶的磷酸化。MAPK的抑制似乎是由c-Raf和B-Raf依赖的MAPK/ERK激酶抑制介导的。多巴胺刺激垂体释放生长激素是通过电压门控钙通道减少细胞内钙离子内流介导的,而非通过抑制腺苷酸环化酶。刺激黑质纹状体通路中的多巴胺D2受体可改善运动障碍患者的肌肉协调活动。麦角林生物碱已被证实对5-HT1和5-HT2血清素受体、D1和D2多巴胺受体以及α-肾上腺素能受体具有显著的亲和力。这可导致多种不同的效应,包括血管收缩、惊厥和幻觉。溴隐亭通过直接刺激纹状体中的多巴胺受体发挥作用。(A2914, A2915, A2916, A2941) |
| 参考文献 |
[1]. Gardner B, et al. Agonist action at D2(long) dopamine receptors: ligand binding and functional assays. Br J Pharmacol. 1998 Jul;124(5):978-84.
[2]. Renodon A, et al. Bromocriptine is a strong inhibitor of brain nitric oxide synthase: possible consequences for the origin of its therapeutic effects.FEBS Lett. 1997 Apr 7;406(1-2):33-6. [3]. Wynalda MA, et al. Assessment of potential interactions between dopamine receptor agonists and various human cytochrome P450 enzymes using a simple in vitro inhibition screen. Drug Metab Dispos. 1997 Oct;25(10):1211-4. [4]. Rana DG, et al. Dopamine mediated antidepressant effect of Mucuna pruriens seeds in various experimental models of depression. Ayu. 2014 Jan;35(1):90-7. [5]. Dieb W, et al. Nigrostriatal dopaminergic depletion increases static orofacial allodynia. J Headache Pain. 2016;17:11 |
| 其他信息 |
药效学
溴隐亭刺激中枢多巴胺能受体,从而产生多种药理作用。目前已鉴定出两种多巴胺能亚家族的五种多巴胺受体。多巴胺D1受体亚家族包括D1和D5亚受体,它们与运动障碍相关。多巴胺D2受体亚家族包括D2、D3和D4亚受体,它们与运动障碍症状的改善相关。因此,D2亚家族受体(主要是D2和D3受体亚型)的特异性激动剂活性是多巴胺能抗帕金森病药物的主要靶点。人们认为,突触后D2受体的激活是多巴胺激动剂发挥抗帕金森病作用的主要原因,而突触前D2受体的激活则具有神经保护作用。这种半合成的麦角衍生物对多巴胺D2受体表现出强效的激动活性。它还表现出对 5-羟色胺 (5-HT)1D、多巴胺 D3、5-HT1A、5-HT2A、5-HT1B 和 5-HT2C 受体的激动剂活性(按结合亲和力递减的顺序排列),对 α2A-肾上腺素能受体、α2C、α2B 和多巴胺 D1 受体的拮抗剂活性,对 5-HT2B 受体的部分激动剂活性,并使多巴胺 D4 和 5-HT7 受体失活。帕金森综合征的发生是由于大脑黑质纹状体通路中约80%的多巴胺能活性丧失。由于纹状体参与调节协调肌肉活动的强度(例如运动、平衡、行走),其活性丧失可能导致肌张力障碍(急性肌肉收缩)、帕金森综合征(包括运动迟缓、震颤、僵硬和情感淡漠等症状)、静坐不能(内心躁动不安)、迟发性运动障碍(通常与长期多巴胺能活性丧失相关的非自主肌肉运动)以及神经阻滞剂恶性综合征,后者发生于黑质纹状体通路多巴胺完全阻断时。大脑中脑边缘通路的多巴胺能活性过高会导致幻觉和妄想;这些多巴胺激动剂的副作用常见于精神分裂症患者,因为他们大脑的这一区域存在过度活跃。多巴胺激动剂的致幻副作用也可能与5-HT2A受体激动作用有关。脑结节漏斗通路起源于下丘脑,终止于垂体。在该通路中,多巴胺抑制垂体前叶泌乳细胞分泌催乳素。结节漏斗通路中多巴胺能活性增强可抑制催乳素分泌,因此溴隐亭是治疗催乳素分泌过多相关疾病的有效药物。肺纤维化可能与溴隐亭对5-HT1B和5-HT2B受体的激动作用有关。 |
| 分子式 |
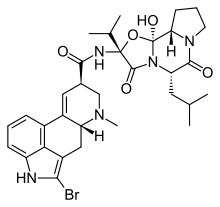
C32H40BRN5O5
|
|---|---|
| 分子量 |
654.606
|
| 精确质量 |
653.221
|
| CAS号 |
25614-03-3
|
| 相关CAS号 |
Bromocriptine mesylate;22260-51-1
|
| PubChem CID |
31101
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.52 g/cm3
|
| 沸点 |
891.3ºC at 760 mmHg
|
| 熔点 |
215-218
|
| 闪点 |
492.8ºC
|
| 蒸汽压 |
4.15E-34mmHg at 25°C
|
| 折射率 |
1.696
|
| LogP |
3.397
|
| tPSA |
118.21
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
43
|
| 分子复杂度/Complexity |
1230
|
| 定义原子立体中心数目 |
6
|
| SMILES |
CC(C[C@H]1C(N2CCC[C@H]2[C@@]3(O)N1C([C@](NC([C@@H]4C=C5C6=C7C(C[C@H]5N(C4)C)=C(NC7=CC=C6)Br)=O)(O3)C(C)C)=O)=O)C
|
| InChi Key |
OZVBMTJYIDMWIL-AYFBDAFISA-N
|
| InChi Code |
InChI=1S/C32H40BrN5O5/c1-16(2)12-24-29(40)37-11-7-10-25(37)32(42)38(24)30(41)31(43-32,17(3)4)35-28(39)18-13-20-19-8-6-9-22-26(19)21(27(33)34-22)14-23(20)36(5)15-18/h6,8-9,13,16-18,23-25,34,42H,7,10-12,14-15H2,1-5H3,(H,35,39)/t18-,23-,24+,25+,31-,32+/m1/s1
|
| 化学名 |
Ergotaman-3',6',18-trione, 2-bromo-12'-hydroxy-2'-(1-methylethyl)-5'-(2-methylpropyl)-, (5'alpha)-
|
| 别名 |
CB154 CB 154 Bromocriptine CB-154
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5276 mL | 7.6381 mL | 15.2763 mL | |
| 5 mM | 0.3055 mL | 1.5276 mL | 3.0553 mL | |
| 10 mM | 0.1528 mL | 0.7638 mL | 1.5276 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
