| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
FEMIB E3 ligase; covalent binding mode targeting cysteine
|
|---|---|
| 体外研究 (In Vitro) |
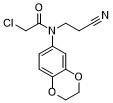
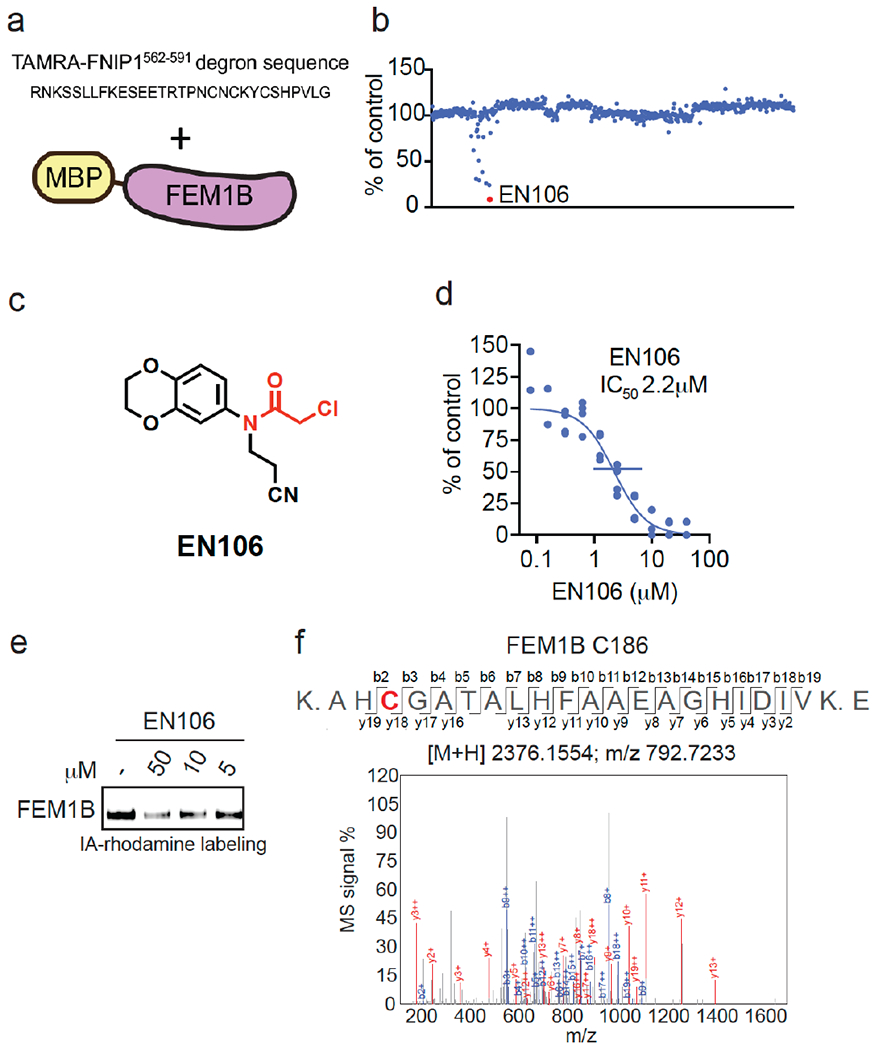
为了鉴定共价FEM1B招募者,我们使用TAMRA偶联的FNIP1562-591 degron和重组小鼠FEM1B在竞争性荧光偏振分析中筛选了566个半胱氨酸反应性共价配体库(图1a-1b,表S1)。通过这一筛选,我们鉴定出氯乙酰胺EN106,其抑制FEM1B-FNIP1 degron荧光偏振的50%抑制浓度(IC50)为2.2μM(图1c-1d)。从最初的筛选来看,EN106对FEM1B与FNIP1去旋器的相互作用显示出最显著的抑制作用(图1b)。EN106显示了与基于凝胶的ABPP用半胱氨酸反应性罗丹明共轭碘乙酰胺(IA罗丹明)探针标记FEM1B的竞争,证实了EN106与FEM1B上的半胱氨酸的直接相互作用(图1e)。通过液相色谱-串联质谱(LC-MS/MS)分析FEM1B胰蛋白酶消化物,对EN106与重组FEM1B的反应性进行分析,结果显示EN106加合物仅存在于C186上,该位点先前被证明对FEM1B底物识别至关重要(图1f)。我们还证明了EN106的非反应性版本NJH-2-082不会抑制FEM1B与FNIP1 degron的相互作用,证实了半胱氨酸反应弹头与C186共价相互作用的重要性(图S1)。[1]
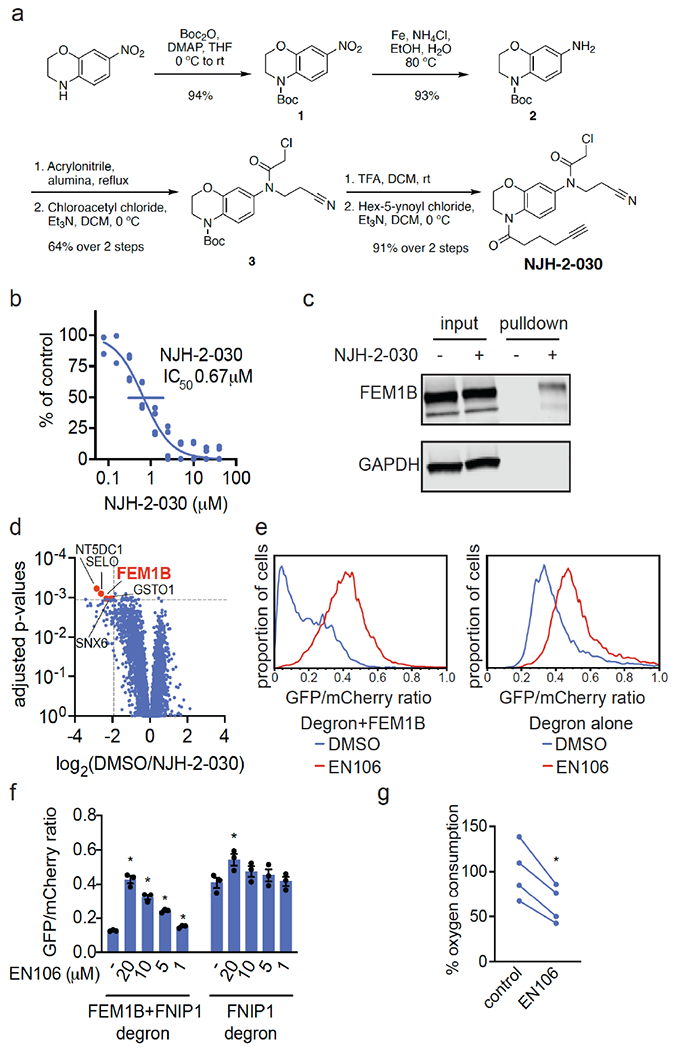
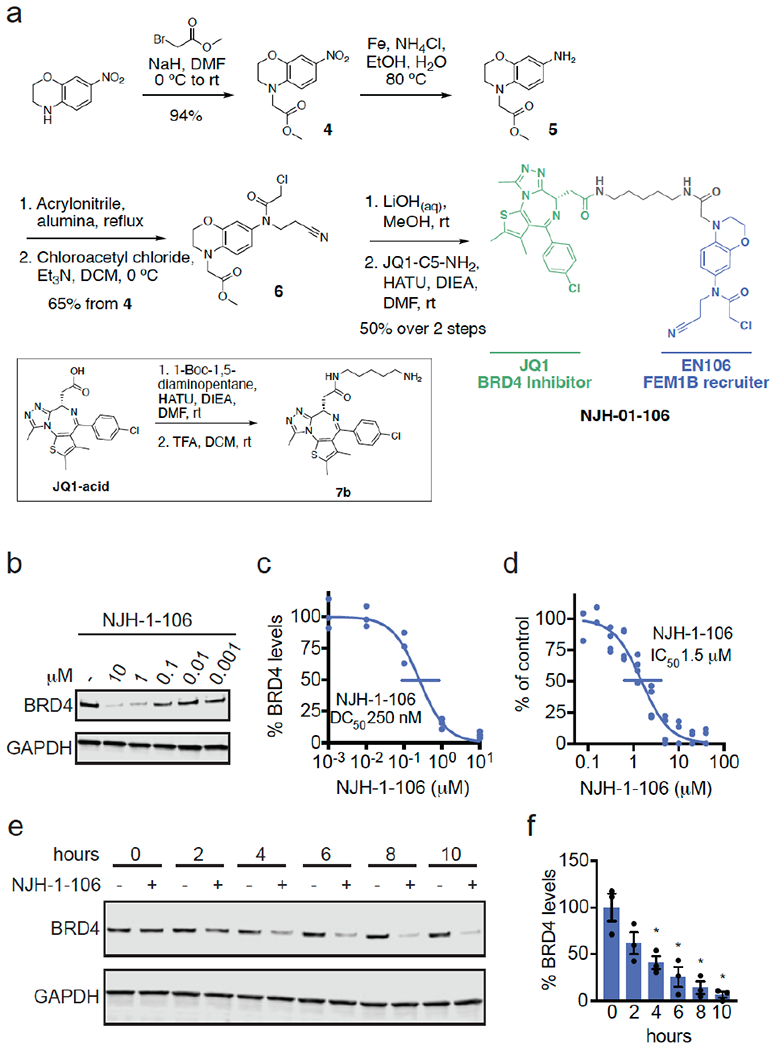
为了确认EN106在细胞中与FEM1B结合,我们合成了NJH-2-030,这是EN106的炔官能化衍生物(图2a)。为了保持C186的接合,通过将苯并二恶烷交换为二氢[1,4]苯并恶嗪支架,将炔烃定位在氯乙酰胺的远端。将起始苯并恶嗪进行Boc保护,得到1,然后还原硝基,得到苯胺2。2与丙烯腈烷基化得到丙腈取代的化合物,将其酰化得到氯乙酰胺3。Boc脱保护和用己-5-炔酰氯酰化提供了炔探针NJH-2-030(图2a)。[1] 为了评估EN106的蛋白质组范围半胱氨酸反应性,我们还进行了竞争性同位素串联正交蛋白水解ABPP(isoTOP ABPP)研究,以定量评估细胞中EN106在蛋白质组范围内的半胱氨酸反应性(图S2;表S2)。虽然我们在化学蛋白质组学实验中没有捕捉到FEM1B,但我们在1465个定量半胱氨酸中只观察到细胞中的两个EN106靶点——HNRNPA3的C63和PRDX3的C127(图S2)。EN106的这些非靶点都不是E3连接酶。鉴于我们在isoTOP ABPP实验中没有检测到FEM1B的C186,我们还在HEK293T细胞中原位使用NJH-2-030探针进行下拉定量蛋白质组学实验,以进一步确认这种紧密的EN106衍生物的靶点参与和蛋白质组范围内的选择性(图2d;表S3)。与DMSO对照组相比,FEM1B是NJH-2-030探针富集最显著的靶标之一,检测到另外四个非靶标——GSTO1、SNX6、SELO和NT5DC1——其中这些蛋白质都不是E3连接酶(图2d;表S3)。这些数据共同表明,EN106或其衍生物在细胞中与FEM1B功能性结合,对其他内源性降解途径成分没有可检测到的脱靶效应。[1] 为了进一步证明EN106破坏了FEM1B在细胞中的底物识别,我们通过流式细胞术监测了与FNIP1 degron连接的GFP的降解,并与HEK293T细胞中同一质粒的IRES驱动的mCherry表达进行了比较EN106处理在FEM1B过表达细胞中以剂量反应方式显著稳定了FNIP1 degron GFP水平,与赋形剂处理的对照组相比(图2e,f)。EN106在缺乏外源表达FEM1B的细胞中增加了FNIP1报告蛋白水平,其程度与之前在缺失FEM1B时观察到的相似,表明该化合物可以靶向内源性E3连接酶(图2e-2f)。EN106不影响E3连接酶cereblon对泊马度胺诱导的无关E4F1 degron的降解(图S3)。因此,这些发现表明EN106不仅参与,而且抑制CUL2FEM1B依赖性泛素化。FNIP1线粒体池的稳定会损害线粒体活性,如耗氧率所示。为了与内源性FEM1B结合并稳定FNIP1,我们发现EN106显著降低了HEK293T细胞中的细胞线粒体耗氧量(图2g)。[1] 为了证明EN106可以在TPD应用中用作共价FEM1B募集剂,我们接下来通过6个不同的连接体将EN106连接到靶向BRD4以及其他BET家族蛋白的BET溴结构域抑制剂JQ1,合成了一系列基于FEM1B的BET溴化结构域降解剂(图3a、图S5)。为了保持炔探针NJH-2-030的核心苯并恶嗪,我们首先连接了一个乙酸酯间隔物以提供甲酯4。硝基被还原,所得苯胺5用丙烯腈单烷基化,并酰化,得到氯乙酰胺中间体6。甲酯在温和的碱性条件下水解,并与具有不同连接体连接的胺7a-7f、JQ1衍生物偶联,得到双功能降解剂NJH-2-088、NJH-1-106、NJH-2-090、NJH-2.091、NJH2-092和NJH-2-02-093(NJH-1-106的方案如图3a所示;图S4)。这些化合物都在HEK293T细胞中不同程度地降解了BRD4,NJH-1-106显示出最佳的降解效力,DC50为250 nM,BRD4的最大降解率为94%(图3b-3c,图S4)。NJH-1-106对FNIP1 degron的FEM1B识别保持抑制活性,IC50为1.5μM(图3d)。这种BRD4降解是时间依赖性的,用NJH-1-106处理4小时后观察到显著降解(图3e-3f)。NJH-106还降解癌症细胞系中的BRD4,包括231MFP乳腺癌症和HAP1白血病癌症细胞系(图S5a–S5b)。[1] 与共价弹头的必要性一致,NJH-1-106、NJH-2-105的非反应性版本没有抑制FEM1B与FNIP1去旋器的相互作用,也没有降解BRD4(图S6a-S6d)。蛋白酶体和NED酰化抑制剂可以减轻BRD4的损失,这与BRD4降解的蛋白酶体和Cullin E3连接酶依赖机制一致(图4a-4b)。NJH-1-106处理HEK293T细胞后,FEM1B水平保持不变(图4a-4b)。用EN106或JQ1预处理细胞也会减弱BRD4的降解,这表明三元复合物和分子两端降解BRD4是必要的(图4c)。此外,与野生型(WT)细胞相比,FEM1B敲除(KO)细胞中的BRD4降解减弱,进一步证明了BRD4的FEM1B依赖性降解(图4d)。我们在FEM1B KO细胞中观察到的不完全拯救可能是由于FEM1B KO群体中残留的野生型细胞或其他潜在的脱靶E3连接酶。用NJH-1-106处理的HEK293T细胞的全球蛋白质组学分析也显示,在4446种定量蛋白质中,BRD4发生了选择性降解,只有大部分未鉴定的PNMAL1是明显的脱靶(图4e;表S4)。然而,PNMAL1可能是我们蛋白质组学分析的假阳性(图S7)。虽然JQ1是一种泛BET溴结构域抑制剂,但我们只观察到BRD4的降解,而没有观察到BRD2或BRD3的降解(表S4)。然而,我们不能排除这些其他溴结构域在较长时间的处理下会被降解。与蛋白质印迹数据相比,观察到的BRD4较小的折叠变化可能反映了基于TMT的定量蛋白质组学中众所周知的折叠变化抑制[1]。 |
| 酶活实验 |
凝胶基ABPP[1]
重组MBP-FEM1B1-377(0.1μg/样品)在25μL PBS中用DMSO载体或EN106或在37°C下预处理30分钟,随后在室温下用IA罗丹明(浓度如图例所示)处理1小时。通过加入4×还原Laemmli-SDS样品加载缓冲液停止反应。在95°C下煮沸5分钟后,将样品在预制的4-20%Criterion TGX凝胶上分离。使用ChemiDoc MP通过凝胶内荧光分析探针标记的蛋白质。 |
| 细胞实验 |
GFP-FNIP1 degron/mHerry的流式细胞术分析[1]
将HEK293T细胞/孔接种到6孔板中。第二天,用0.1μg pCS2-GFP-FNIP1562-591-IRES-mCherry或0.1μg的pCS2-E4F123432-GFP-IRES-mCherry转染细胞,如图所示,用0.075μg的pCS2-3xFLAG-FM1B转染。每次转染时,将空的pCS2加入到含有12μg聚乙烯亚胺(PEI)的300μl Opti-MEM中的2μg总DNA中。用65μl转染混合物转染每个孔。转染后12小时,加入指示浓度的EN106或DMSO。经过12小时的EN106处理后,细胞被胰蛋白酶消化,离心,重新悬浮在DMEM+10%FBS中,并在Fortessa X20上进行分析。使用FlowJo处理数据,所有定量均为GFP/mHerry比值的中位数。对于泊马度胺处理的细胞,在分析前加入10μM泊马度酰胺4小时。 |
| 参考文献 | |
| 其他信息 |
蛋白水解靶向嵌合体(PROTACs)是一种异双功能化合物,由蛋白靶向配体与E3连接酶募集剂连接而成,已成为靶向蛋白降解(TPD)的一种强效治疗手段。尽管TPD方法在药物研发中应用广泛,但针对人类细胞中存在的600多种E3连接酶,目前可用的E3连接酶募集剂却寥寥无几。本文中,我们发现了一种半胱氨酸反应性共价配体EN106,它靶向FEM1B,一种近期被发现是细胞应对还原应激的关键组分的E3连接酶。EN106通过靶向FEM1B中的C186位点,干扰FEM1B对其关键还原应激底物FNIP1的识别。我们进一步证实,EN106 可作为 FEM1B 的共价募集剂用于 TPD 应用。我们证明,将 EN106 与 BET 溴结构域抑制剂 JQ1 或激酶抑制剂达沙替尼连接的 PROTAC 分别可导致 BRD4 和 BCR-ABL 的降解。我们的研究展示了一种靶向天然 E3 连接酶-底物结合位点的共价配体,并强调了共价配体筛选在扩展适用于 TPD 应用的 E3 连接酶募集剂库方面的实用性。[1]
在本研究中,我们发现了针对 FEM1B(一种控制还原应激反应的 E3 连接酶)的共价募集剂 EN106,该募集剂可用于 TPD 应用。虽然锌已被发现是一种将 FEM1B 与其内源性底物 FNIP1 连接在一起的分子胶,但 EN106 是第一个针对 FEM1B 的合成小分子配体。 EN106 是一种早期先导化合物,对 FEM1B 的抑制效力为低微摩尔级,其氯乙酰胺活性基团代谢不稳定,需要进一步的药物化学研究来提高其效力、选择性和类药性。我们证实 EN106 靶向 FEM1B 中一个对底物识别至关重要的半胱氨酸残基。我们发现 EN106 可作为双功能降解剂中的 FEM1B 募集剂,将潜在的新底物募集到 FEM1B 的生理靶点识别位点,从而使其以最佳方式通过 CUL2-RBX1 进行泛素化。令人惊讶的是,我们观察到所有基于 FEM1B 的 BRD4 PROTAC 均能降解 BRD4,且与连接子长度无关,尽管存在对最佳 Dmax 和 DC50 值的偏好。这些数据可能表明,只要避免蛋白质碰撞,共价 FEM1B 降解剂的正协同性并非至关重要。未来的研究需要确定EN106或其衍生物是否能作为分子胶降解剂,募集潜在的新底物进行FEM1B依赖的泛素化和降解。更广泛地说,未来研究还应关注能否开发细胞状态特异性降解剂。此外,确定EN106及其更有效的衍生物是否可用于治疗,在某些癌症情况下通过稳定FNIP1来抑制CUL2FEM1B并破坏还原应激信号通路,也是未来的研究方向。总而言之,我们的研究强调了共价配体筛选在拓展TPD应用中E3连接酶募集剂范围方面的实用性。[1] |
| 分子式 |
C13H13CLN2O3
|
|---|---|
| 分子量 |
280.706922292709
|
| 精确质量 |
280.06
|
| 元素分析 |
C, 55.62; H, 4.67; Cl, 12.63; N, 9.98; O, 17.10
|
| CAS号 |
757192-67-9
|
| PubChem CID |
5100616
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.351±0.06 g/cm3(Predicted)
|
| 沸点 |
489.1±45.0 °C(Predicted)
|
| LogP |
1.3
|
| tPSA |
62.6
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
19
|
| 分子复杂度/Complexity |
368
|
| 定义原子立体中心数目 |
0
|
| SMILES |
ClCC(N(CCC#N)C1C=CC2=C(C=1)OCCO2)=O
|
| InChi Key |
GLDJSVHDOXCYPT-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C13H13ClN2O3/c14-9-13(17)16(5-1-4-15)10-2-3-11-12(8-10)19-7-6-18-11/h2-3,8H,1,5-7,9H2
|
| 化学名 |
2-chloro-N-(2-cyanoethyl)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)acetamide
|
| 别名 |
EN106; EN-106; 757192-67-9; 2-chloro-N-(2-cyanoethyl)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)acetamide; 2-chloro-N-(2-cyanoethyl)-N-2,3-dihydro-1,4-benzodioxin-6-ylacetamide; CHEMBL5289726; SCHEMBL23804991; DTXSID001152090; EN 106
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~250 mg/mL (~890.60 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5624 mL | 17.8120 mL | 35.6240 mL | |
| 5 mM | 0.7125 mL | 3.5624 mL | 7.1248 mL | |
| 10 mM | 0.3562 mL | 1.7812 mL | 3.5624 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
