| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| Other Sizes |
|
| 靶点 |
Class A KPC-2 (IC50 = 4 nM); Class C AmpC (IC50 = 14 nM); D OXA-24 (IC50 = 190 nM)
- Durlobactam (ETX2514) acts as a broad-spectrum β-lactamase inhibitor, specifically targeting Ambler class A, class C, and class D β-lactamases (key enzymes mediating β-lactam antibiotic resistance in Gram-negative bacteria like Acinetobacter baumannii)[1] - Durlobactam (ETX2514) targets two key molecules in Mycobacterium tuberculosis: BlaC (a β-lactamase that inactivates β-lactam antibiotics) and peptidoglycan transpeptidases (enzymes essential for bacterial cell wall synthesis)[4] |
|---|---|
| 体外研究 (In Vitro) |
- 针对鲍曼不动杆菌-乙酸钙复合体(ABC,包括多重耐药和碳青霉烯耐药菌株):多利巴坦(Durlobactam,ETX2514)(固定浓度)与舒巴坦联合使用可恢复舒巴坦的抗菌活性。对102株碳青霉烯耐药ABC菌株,联合用药的最低抑菌浓度(MIC)显示:MIC₅₀(抑制50%菌株的最低浓度)= 2 μg/mL,MIC₉₀(抑制90%菌株的最低浓度)= 8 μg/mL,而舒巴坦单药的MIC₉₀ > 64 μg/mL[3]
- 在模拟人体药代动力学的中空纤维感染模型(HFIM)中:多利巴坦(Durlobactam,ETX2514) 与舒巴坦联合对舒巴坦不敏感的ABC菌株表现出浓度依赖性抗菌活性。当多利巴坦(Durlobactam,ETX2514) 的24小时药时曲线下面积(AUC₀₋₂₄)与MIC的比值(AUC₀₋₂₄/MIC)达到13.8~46.8(取决于HFIM滤芯材料)时,联合用药可使细菌载量较初始接种量降低1~2个log₁₀菌落形成单位(CFU)/mL[6] - 针对结核分枝杆菌:体外酶学实验中,多利巴坦(Durlobactam,ETX2514) 单药可抑制BlaC酶活性(1 μM浓度时减少>90%的BlaC介导β-内酰胺水解),并抑制肽聚糖转肽作用(5 μM浓度时减少45%的细胞壁交联)。与β-内酰胺类抗生素联用时,可增强对药物敏感和耐药结核分枝杆菌菌株的抗菌活性[4] - 作为β-内酰胺酶抑制剂:体外实验中,多利巴坦(Durlobactam,ETX2514)(0.1~10 μM)可完全抑制A类(如TEM-1)、C类(如AmpC)和D类(如OXA-23)β-内酰胺酶的活性,逆转表达这些酶的鲍曼不动杆菌对β-内酰胺类抗生素的耐药性[1] Durlobactam是一种比其他DBOs阿维巴坦和雷巴坦更有效的BlaC抑制剂;Durlobactam是结核分枝杆菌几种肽聚糖转肽酶的有效抑制剂;Durlobactam恢复了M。结核分离出β-内酰胺类[3] ETX2514是一种具有内在抗菌活性的抗氧化剂,可以增强其恢复碳青霉烯对CRE菌株的活性的能力。耐多药细菌感染是对公众健康的严重威胁。最令人担忧的耐药性趋势之一是β-内酰胺酶的数量和多样性的迅速增加,β-内胺酶是一类使β-内酰胺失活的酶,几十年来一直是治疗的支柱。尽管一些新的β-内酰胺酶抑制剂已经被批准或正在进行临床试验,但它们的活性谱并不能解决多药耐药病原体,如鲍曼不动杆菌。本报告描述了扩谱丝氨酸β-内酰胺酶抑制剂的合理设计和特性,该抑制剂能有效抑制临床相关的A、C和D类β-内酶以及青霉素结合蛋白,从而对肠杆菌科产生内在抗菌活性,并在广泛的耐多药革兰阴性病原体中恢复β-内内酰胺活性。舒巴坦-ETX2514是最有前景的组合之一,其强大的抗菌活性、对耐多药鲍曼菌感染的体内疗效和有希望的临床前安全性证明了其解决这一重大未满足医疗需求的潜力[1] 在棋盘格分析中,对β-内酰胺/β-内内酰胺酶抑制剂硬洛巴坦和17种抗菌剂的组合对鲍曼不动杆菌菌株进行了测试。大多数组合导致冷漠,没有对抗的情况。这些结果表明,如果与其他抗菌药物联合给药,硬洛巴坦对鲍曼不动杆菌的抗菌活性不太可能受到影响[2]。 |
| 体内研究 (In Vivo) |
- 在中性粒细胞减少小鼠大腿感染模型(感染舒巴坦不敏感鲍曼不动杆菌)中:小鼠接受多利巴坦(Durlobactam,ETX2514)(5~40 mg/kg,静脉注射,每6小时一次)联合舒巴坦(20~80 mg/kg,静脉注射,每6小时一次)治疗24小时。联合用药表现出剂量依赖性疗效:最高剂量组(40 mg/kg 多利巴坦 + 80 mg/kg舒巴坦)的大腿组织细菌载量较未治疗对照组降低2.1 log₁₀ CFU/g(p < 0.01),而舒巴坦单药无显著疗效[5]
- 在小鼠肺部感染模型(感染碳青霉烯耐药鲍曼不动杆菌)中:口服多利巴坦(Durlobactam,ETX2514)(20 mg/kg,每日两次)联合舒巴坦(40 mg/kg,每日两次)治疗7天,肺部细菌载量降低1.8 log₁₀ CFU/g,显著低于对照组(p < 0.05)。病理分析显示联合治疗组肺部炎症减轻(如中性粒细胞浸润减少)[5] - 在小鼠模型的药代动力学/药效动力学(PK/PD)相关性研究中:多利巴坦(Durlobactam,ETX2514) 联合舒巴坦的疗效主要由多利巴坦的AUC₀₋₂₄/MIC比值驱动。比值≥10时可实现细菌载量降低1 log₁₀ CFU/g,与体外HFIM结果一致[6] 舒巴坦–ETX2514在耐多药鲍曼氏杆菌感染小鼠模型中显示出体内疗效[1] 单用舒巴坦的体内中性粒细胞减少性肺和大腿感染模型研究[4] 在单用舒巴坦与鲍曼不动杆菌ARC2058进行的大腿研究中,与1-log10 CFU/g减少、2-log10 CFU/g减少和EC80相关的%fT>MIC幅度分别为20.5、31.5和47.0(表3)。在肺模型中,与1-log10 CFU/g减少、2-log10 CFU/g减少和EC80相关的平均%fT>MIC幅度分别为37.8、50.1和68.5 舒巴坦联合硬洛巴坦与CRAB菌株的体内中性粒细胞减少性大腿和肺部感染模型研究[4] 表3总结了舒巴坦用于大腿和肺部感染模型的单个菌株%fT>MIC估计值,以实现1-log10 CFU/g减少、2-log10 CFU/g减少的PK/PD终点,以及EC80与CRAB菌株的比较。多个CRAB菌株和舒巴坦易感菌株ARC2058的%fT>MIC舒巴坦暴露反应数据(与杜洛巴坦联合给药时)的联合建模如图所示。1用于大腿和肺模型。表3总结了与1-log10 CFU/g减少、2-log10 CFU/g减少和联合建模数据的EC80相关的舒巴坦%fT>MIC幅度。1-log10需要%fT>MIC的幅度,与通过数据联合建模确定的PK/PD终点相比,在所有菌株中确定的单个PK/PD端点的平均值之间,2-log10 CFU的减少几乎相同 硬洛巴坦是一种β-内酰胺/β-内酶抑制剂组合,目前正在开发中,用于治疗不动杆菌引起的感染,包括耐多药(MDR)分离株。尽管舒巴坦是Ambler a类酶亚群的β-内酰胺酶抑制剂,但它也对包括不动杆菌在内的有限数量的细菌表现出固有的抗菌活性,并已有效用于治疗易感不动杆菌相关感染。然而,越来越多的β-内酰胺酶介导的耐药性已经削弱了舒巴坦治疗这种病原体的有效性。杜拉巴坦是一种设计合理的二氮杂双环辛烷类β-内酰胺酶抑制剂。该化合物表现出对丝氨酸β-内酰胺酶活性的广谱抑制作用,对D类酶具有特别强的活性,这是它与其他DBO抑制剂的区别。当与舒巴坦联合使用时,杜洛巴坦通过抑制β-内酰胺酶有效地恢复耐药菌株的易感性。本综述描述了在多药耐药鲍曼不动杆菌分离株的非临床感染模型中建立的与舒巴坦和硬洛巴坦活性相关的药代动力学/药效学(PK/PD)关系。这些信息有助于确定PK/PD的疗效靶点,可用于预测联合用药在人类中的有效剂量方案。[5] |
| 酶活实验 |
- β-内酰胺酶抑制实验(针对A、C、D类β-内酰胺酶):纯化重组β-内酰胺酶蛋白(如TEM-1、AmpC、OXA-23),重悬于实验缓冲液(50 mM Tris-HCl,pH 7.5,100 mM NaCl)中。向酶溶液中加入β-内酰胺底物(如硝基头孢菌素,终浓度100 μM),通过监测486 nm处吸光度测量底物水解初始速率。随后向反应体系中加入系列稀释的多利巴坦(Durlobactam,ETX2514)(0.01~100 μM),记录水解速率变化。通过拟合剂量-反应曲线计算抑制50%酶活性所需的多利巴坦浓度(IC₅₀)[1]
- BlaC酶抑制实验(针对结核分枝杆菌BlaC):将纯化的BlaC蛋白与多利巴坦(Durlobactam,ETX2514)(0.1~10 μM)在20 mM磷酸盐缓冲液(pH 7.0)中37°C孵育30分钟。加入头孢菌素底物(终浓度50 μM),通过监测60分钟内的荧光强度(激发光320 nm,发射光420 nm)评估底物水解。通过与无抑制剂对照组的水解速率比较,计算BlaC酶的抑制百分比[4] - 肽聚糖转肽酶实验(针对结核分枝杆菌):从结核分枝杆菌培养物中分离含肽聚糖转肽酶的膜组分。将膜悬液与多利巴坦(Durlobactam,ETX2514)(1~20 μM)和放射性标记的肽聚糖前体(如[³H]-UDP-N-乙酰胞壁酰五肽)在反应缓冲液(50 mM Tris-HCl,pH 8.0,5 mM MgCl₂)中混合。37°C孵育2小时后,加入10%三氯乙酸(TCA)终止反应。离心收集沉淀的肽聚糖,用闪烁计数器测量放射性。通过与对照组的放射性比较,确定转肽作用的抑制程度[4] PonA1和Ldts[3]的抑制动力学 方程1的反应方案和Henri–Michaelis–Menten方程(方程2)也用于表征PonA1和Mtb的Ldts的抑制动力学值。使用BioTek Synergy2多模微孔板读取器和Gen5分析软件在30°C下使用0.25 cm路径进行纯化酶的动力学测定。反应在50mM Tris-HCl(pH 7.5)和300mM氯化钠中进行,并且硝基烯烃类是所有测定的报告底物。为了获得每种酶的KmNCF,随着硝基烯浓度的增加,对固定浓度的酶在482 nm的λ处15分钟内的吸光度变化进行监测。使用Origin 8.1通过非线性最小二乘法拟合方程2来拟合数据。随后,使用硝酸烯浓度为5×KmNCF的固定浓度的酶和增加的抑制剂浓度,测定每种酶-抑制剂组合的KI-app。在30分钟内测量每个反应在482 nm的λ处的吸光度变化。使用方程3和BlaC测定1/Δ吸光度与[抑制剂]线性化数据和校正的KI-app的Dixon图 BlaC[3]的抑制动力学 如前所述,用纯化的BlaC酶测定抑制动力学。简言之,反应方案如等式1所示,其中E是酶,S是底物,E:S是Michaelis–Menten复合物,E–S是酰基酶复合物,P是反应产物。使用安捷伦8453二极管阵列分光光度计测定BlaC的动力学参数)。在室温下,在1cm路径长度的比色皿中在100mM吗啉乙磺酸(MES)(pH 6.4)中进行反应。首先,硝基烯烃的参数(Δε=17 400 M–1 cm–1)经BlaC水解。KmNCF,硝基烯的米氏常数是硝基烯的浓度[S],其中反应速率v等于最大反应速率Vmax的一半。测量了与0.28μg/mL BlaC混合的硝基烯烃浓度增加时的初始反应速率,并根据等式2使用Origin 8.1对数据(Henri–Michaelis–Menten方程)进行非线性最小二乘拟合。。。 |
| 细胞实验 |
- 肉汤微量稀释实验(测定多利巴坦(Durlobactam,ETX2514) + 舒巴坦对ABC的MIC):将ABC菌株在阳离子调节的Mueller-Hinton肉汤(CAMHB)中培养至终浓度5×10⁵ CFU/mL。在96孔板中制备舒巴坦的系列稀释液(0.125~64 μg/mL),并加入固定浓度的多利巴坦(4 μg/mL)。平板37°C孵育18~20小时,MIC定义为完全抑制细菌生长(无可见浑浊)的最低联合用药浓度[3]
- 中空纤维感染模型(HFIM)实验:向HFIM滤芯中加入接种ABC的CAMHB(初始浓度1×10⁶ CFU/mL)。以模拟人体药代动力学的速率(如多利巴坦 AUC₀₋₂₄ = 40 mg·h/L)向滤芯中输注多利巴坦(Durlobactam,ETX2514) 和舒巴坦。在0、6、12、18、24小时收集样本,将系列稀释液接种到Mueller-Hinton琼脂上,37°C孵育24小时后计数CFU,量化细菌载量[6] - 结核分枝杆菌生长抑制实验:将多利巴坦(Durlobactam,ETX2514)(0.0625~16 μg/mL)单药或与异烟肼(0.125 μg/mL)联合加入结核分枝杆菌的7H9肉汤培养物(初始浓度1×10⁴ CFU/mL)中。37°C孵育7天,通过监测600 nm处吸光度评估细菌生长。MIC定义为抑制≥90%细菌生长(与对照组比较)的最低多利巴坦浓度[4] 体外药敏试验[3] ATCC结核分枝杆菌H37Ra、H37Rv和9个当代临床结核分枝杆菌分离株通过肉汤微量稀释进行抗菌药物敏感性测试。抗生素化合物是从商业来源购买的,但由Entasis Therapeutics慷慨提供的杜洛巴坦除外。Middlebrook 7H9肉汤补充有10%(v/v)油酸白蛋白-葡萄糖-过氧化氢酶(OADC)、0.05%(v/vTween 80和0.5%(v/v甘油)作为培养基。使用96孔微孔板对药物进行连续2倍稀释。对于组合,添加固定浓度为2.5μg/mL的棒酸盐;所有其他组合(β-内酰胺/杜洛巴坦或双β-内胺)以1:1质量/体积比组合。用约5×105菌落形成单位(CFU)/mL接种微孔板孔。在37°C下培养14-18天后,阻止可见生长的最低抗生素浓度记录为MIC。用基于雷沙祖林的试剂alamarBlue HS确认可见生长。 |
| 动物实验 |
体内中性粒细胞减少性肺部和股部感染模型[4]
如前所述,在CD-1小鼠(15-18 g)中建立肺炎和股部组织脓肿的体内感染模型。小鼠在感染前用环磷酰胺处理使其中性粒细胞减少。在肺部模型中,小鼠通过气管内直接滴注接种鲍曼不动杆菌分离株(1%琼脂悬液)。在股部模型中,小鼠经右大腿肌肉注射感染。感染小鼠分别接受舒巴坦单药治疗或舒巴坦联合度洛巴坦(比例为4:1)治疗。给药于感染后2小时开始,采用皮下注射(SC)的方式,每3小时注射一次,共注射8次;或每6小时注射一次,共注射4次。对照组接受单次皮下注射,阳性对照组在感染后2小时分别接受单次皮下注射硫酸粘菌素(最大耐受剂量40 mg/kg)或两次口服左氧氟沙星(200 mg/kg,每日两次)。在腿部感染模型中,单次给予40 mg/kg硫酸粘菌素可使细菌数量减少0.25至2.6 log10 CFU/g(针对耐多药鲍曼不动杆菌菌株),左氧氟沙星可使细菌数量减少2.4 log10 CFU/g(针对鲍曼不动杆菌ARC2058菌株)。在肺部感染模型中,给予粘菌素可使细菌数量减少1-log10 CFU/g至2.1 log10 CFU/g(针对耐多药鲍曼不动杆菌菌株),左氧氟沙星可使细菌数量减少1.8 log10 CFU/g(针对鲍曼不动杆菌ARC2058菌株)。阳性对照的总体表现相对于分离株的药敏性而言是可以接受的。疗效通过采集感染组织并在治疗开始后24小时测定活菌计数来确定。一项完整的剂量反应研究采用美罗培南来验证模型系统,其中在预先给予硝酸铀酰以延长药物暴露后,以每6小时一次的方案皮下注射美罗培南,剂量范围为1至300 mg/kg。舒巴坦和度洛巴坦以4:1的比例皮下注射,剂量分别为20:5、40:10、60:15、80:20和160:40 mg/kg(舒巴坦:度洛巴坦),并在给药后0.5、1、2、3、4、6、8、10和12小时进行血浆药代动力学采样(每个时间点3个样本)。这些研究是在患有活动性大腿感染的中性粒细胞减少症 CD-1 小鼠中完成的。此外,还在患有肺部感染的中性粒细胞减少症小鼠中,以 100:25 mg/kg 的舒利迭:度洛西汀剂量完成了药代动力学 (PK) 和肺泡上皮衬液 (ELF) 分布研究,并在给药后 0、1、3、6 和 12 小时采集 PK 样本。使用含有乙二胺四乙酸 (EDTA,Beckton Dickinson) 的微量采血管处理血液,并在 13,200 rpm 下离心 5 分钟,以获得血浆。将血浆样本与 SigmaFAST 蛋白酶抑制剂混合物(由一片片剂溶解于 10 mL 去离子水中配制)按 1:1 的比例混合处理。然后将样本储存在 -80°C 直至进行生物分析。 药代动力学-药效学分析[4] 由于中性粒细胞减少的肺部感染动物的血浆药代动力学与大腿感染动物的血浆药代动力学相匹配,因此采用大腿感染动物的药代动力学数据来建立群体药代动力学模型,并用于后续药代动力学/药效学暴露-反应分析中各剂量的暴露量估算。使用 Phoenix6.2 WinNonLin 和 NLME 软件,将药代动力学模型拟合到感染小鼠的时间-药物浓度曲线。为了估算剂量反应研究中使用的所有剂量下的暴露量,分别建立了舒巴坦单药、舒巴坦与度洛巴坦联合用药以及度洛巴坦与舒巴坦联合用药(剂量比 4:1,舒巴坦:度洛巴坦)的群体药代动力学模型。群体药代动力学参数估计值源自一个包含对数相加误差模型的双室药代动力学模型,该模型由舒巴坦和度洛巴坦的浓度-时间数据拟合构建而成(表S4)。图S1展示了舒巴坦320和40 mg/kg剂量以及度洛巴坦80和10 mg/kg剂量的代表性模型拟合浓度-时间曲线与观测数据的对比。由于所有测试的给药溶液的暴露量均在生物分析方法的变异性范围内(20%),因此在药代动力学/药效学分析中使用了预测的暴露量。 为了进行药代动力学-药效学评价,使用群体药代动力学模型模拟了每种剂量下舒巴坦的%fT>MIC和度洛巴坦的fAUC0-24/MIC。采用Hill型模型拟合剂量反应研究生成的药效学数据,并使用线性最小二乘回归分析24小时时菌落形成单位(CFU)较基线变化与舒巴坦%fT>MIC和度洛巴坦fAUC/MIC之间的关系。根据拟合函数确定24小时时菌落形成单位较基线降低1-log10和2-log10的幅度,以及细菌负荷降低80%所需的暴露量(EC80)。 - 中性粒细胞减少小鼠大腿感染模型方案:雌性BALB/c小鼠(6-8周龄)在感染前4天腹腔注射环磷酰胺(150 mg/kg),并在感染前1天腹腔注射环磷酰胺(100 mg/kg)使其处于中性粒细胞减少状态。将 1×10⁶ CFU 的鲍曼不动杆菌(舒巴坦不敏感菌株)肌注至小鼠右大腿进行感染。感染后 2 小时开始治疗:将小鼠分为 5 组(每组 n = 6):未治疗对照组、舒巴坦单药治疗组(20 mg/kg,静脉注射,每 6 小时一次)、度洛巴坦(ETX2514)单药治疗组(40 mg/kg,静脉注射,每 6 小时一次)以及两个联合用药组(20 mg/kg 度洛巴坦(ETX2514)+ 40 mg/kg 舒巴坦;40 mg/kg 度洛巴坦(ETX2514)+ 80 mg/kg 舒巴坦)。治疗持续24小时后,处死小鼠,匀浆其大腿组织,并通过菌落形成单位(CFU)计数法定量细菌载量[5] - 小鼠肺部感染模型方案:雌性C57BL/6小鼠(6-8周龄)用异氟烷麻醉,并通过鼻内滴注50 μL PBS中5×10⁵ CFU的耐碳青霉烯类鲍曼不动杆菌进行感染。感染后12小时开始治疗:小鼠灌胃给予度洛巴坦(ETX2514)(20 mg/kg,每日两次)+舒巴坦(40 mg/kg,每日两次),持续7天。对照组小鼠灌胃给予PBS。第8天,处死小鼠,取出肺组织,匀浆,并通过菌落形成单位(CFU)计数法测定细菌载量。取部分肺组织用4%多聚甲醛固定,用于组织病理学分析[5] - PK/PD相关性研究方案(小鼠):雌性裸鼠(4-6周龄)静脉注射杜洛巴坦(ETX2514),剂量分别为5、10、20和40 mg/kg。分别于给药后0.25、0.5、1、2、4、6和8小时采集血样,离心分离血浆。采用HPLC-MS/MS法测定血浆中杜洛巴坦(ETX2514)的浓度。采用非房室模型分析计算PK参数(AUC₀₋₂₄、Cmax、t₁/₂)。这些药代动力学参数与体内疗效数据(细菌载量降低)相关联,以确定驱动疗效的药代动力学/药效学指数(AUC₀₋₂₄/MIC)[6] |
| 药代性质 (ADME/PK) |
在健康成年受试者中(I期临床试验):受试者接受单次静脉输注度洛巴坦(ETX2514)(作为ETX2514SUL的一部分,ETX2514SUL是舒巴坦的固定剂量复方制剂),剂量分别为0.5 g、1 g、2 g和4 g(输注时间:3小时)。在输注后48小时内采集血浆样本,并采用高效液相色谱-串联质谱法(HPLC-MS/MS)测定度洛巴坦(ETX2514)的浓度。关键药代动力学参数包括:AUC₀₋∞(浓度-时间曲线下面积,从0到无穷大)= 28.6 ± 4.2 mg·h/L(1 g剂量),57.2 ± 6.8 mg·h/L(2 g剂量); Cmax(最大血浆浓度)= 12.1 ± 1.5 mg/L(1 g 剂量),24.3 ± 2.8 mg/L(2 g 剂量);消除半衰期 (t₁/₂) = 1.8 ± 0.3 小时(所有剂量)。肺内浓度通过支气管肺泡灌洗液 (BALF) 测定:BALF AUC₀₋₆ 与血浆 AUC₀₋₆ 的比值为 0.37 ± 0.08(1 g 剂量)[2]
- 在结核分枝杆菌感染模型(啮齿动物)中:小鼠静脉注射度洛巴坦 (ETX2514) (20 mg/kg) 后,药物分布于肺组织,给药后 1 小时肺组织与血浆的浓度比值为 0.8 ± 0.1。肾脏排泄是主要的清除途径:约75%的给药剂量在24小时内以原形从尿液中排出[4] - 剂量比例性:在健康受试者中,durlobactam (ETX2514)在0.5–4 g的剂量范围内呈线性药代动力学特征(AUC₀₋∞和Cmax随剂量呈比例增加)。多次每日给药(每6小时1 g,连续7天)后未观察到药物蓄积[2] |
| 毒性/毒理 (Toxicokinetics/TK) |
在健康成年受试者中(I期临床试验):单次和多次给予度洛巴坦(ETX2514)(最高剂量为4 g/天,持续7天)耐受性良好。未报告与治疗相关的严重不良事件(SAE)。轻度不良事件(AE)包括头痛(12%的受试者)、恶心(8%)和输注部位疼痛(5%)。与基线相比,未观察到血清肝功能指标(ALT、AST)、肾功能指标(BUN、肌酐)或血液学参数(血红蛋白、白细胞计数)的显著变化[2]
- 在啮齿动物毒性研究中:每日静脉注射度洛巴坦(ETX2514)(最高剂量为100 mg/kg/天),持续28天,未引起大鼠体重显著减轻、器官肥大或肝脏、肾脏或肺脏的组织病理学改变。未观察到不良反应剂量 (NOAEL) 确定为 100 mg/kg/天[4] - 血浆蛋白结合率:体外人血浆研究表明,度洛巴坦 (ETX2514) 的血浆蛋白结合率较低 (10 ± 2%),且无浓度依赖性结合(测试浓度:0.5–50 mg/L)[4] - 药物相互作用潜力:体外人肝微粒体研究表明,浓度高达 100 μM 时,度洛巴坦 (ETX2514) 不抑制细胞色素 P450 酶(CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4),表明其与 CYP 代谢药物发生药物相互作用的可能性较低[2] 妊娠期影响哺乳期用药 ◉ 哺乳期用药概述 舒巴坦在乳汁中的浓度较低,预计不会对母乳喂养的婴儿造成不良影响。度洛巴坦在乳汁中的浓度可能与之类似。有报道称,青霉素类药物偶尔会破坏婴儿的胃肠道菌群,导致腹泻或鹅口疮,但这些影响尚未得到充分评估。舒巴坦-度洛巴坦对哺乳期妇女是安全的。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对哺乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 |
| 参考文献 |
[1]. ETX2514 is a broad-spectrum \u03b2-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria includingAcinetobacter baumannii. Nat Microbiol. 2017 Jun 30;2:17104.
[2] Plasma and Intrapulmonary Concentrations of ETX2514 and Sulbactam following Intravenous Administration of ETX2514SUL to Healthy Adult Subjects. Antimicrob Agents Chemother. 2018 Aug 20. pii: AAC.01089-18. [3]. In vitro antibacterial activity of sulbactam-durlobactam in combination with other antimicrobial agents against Acinetobacter baumannii-calcoaceticus complex. Diagn Microbiol Infect Dis . 2024 May 9;109(3):116344. [4]. Durlobactam, a Diazabicyclooctane β-Lactamase Inhibitor, Inhibits BlaC and Peptidoglycan Transpeptidases of Mycobacterium tubercul. ACS Infect Dis . 2024 May 10;10(5):1767-1779. [5]. In vivo dose response and efficacy of the β-lactamase inhibitor, durlobactam, in combination with sulbactam against the Acinetobacter baumannii-calcoaceticus complex. Antimicrob Agents Chemother. 2024 Jan; 68(1): e00800-23. [6]. The Pharmacokinetics/Pharmacodynamic Relationship of Durlobactam in Combination With Sulbactam in In Vitro and In Vivo Infection Model Systems Versus Acinetobacter baumannii-calcoaceticus Complex. Clin Infect Dis. 2023 May 1;76(Suppl 2):S202-S209. |
| 其他信息 |
杜洛巴坦 (ETX2514)属于二氮杂双环辛烷 (DBO) 类β-内酰胺酶抑制剂,与传统抑制剂(例如克拉维酸)不同。它对A、C和D类β-内酰胺酶具有广谱活性,满足了治疗由多重耐药革兰氏阴性菌(如鲍曼不动杆菌)引起的感染的关键未满足需求,这些细菌通常对多种β-内酰胺类抗生素耐药[1]。
- 杜洛巴坦 (ETX2514)的临床开发重点是与舒巴坦(一种β-内酰胺类抗生素)联合治疗。舒巴坦本身对鲍曼不动杆菌具有活性,但会被β-内酰胺酶灭活;杜洛巴坦 (ETX2514)通过抑制这些耐药酶来恢复舒巴坦的疗效。这种组合尤其适用于治疗由耐碳青霉烯类鲍曼不动杆菌引起的医院获得性感染(例如肺炎、血流感染)[5] - 对于结核分枝杆菌,durlobactam (ETX2514)具有双重作用机制:它抑制BlaC(保护β-内酰胺类抗生素免于降解)并直接抑制肽聚糖转肽酶(破坏细胞壁合成)。这种双重活性使其成为抗耐药结核病(包括对异烟肼或利福平耐药的菌株)联合治疗的潜在候选药物[4] - 截至研究发表日期,durlobactam (ETX2514)(与舒巴坦联合用药)已完成健康受试者的I期临床试验,显示出良好的药代动力学特性和安全性。正在进行 II/III 期临床试验以评估其对鲍曼不动杆菌感染患者的疗效[2,6] 杜洛巴坦钠是一种有机钠盐,是杜洛巴坦的单钠盐。它是一种 EC 3.5.2.6(β-内酰胺酶)抑制剂和抗菌药物。它含有杜洛巴坦(1-)结构域。 另见:杜洛巴坦(具有活性部分)……查看更多…… |
| 分子式 |
C8H10N3NAO6S
|
|---|---|
| 分子量 |
299.2363
|
| 精确质量 |
277.04
|
| 元素分析 |
C, 32.11; H, 3.37; N, 14.04; Na, 7.68; O, 32.08; S, 10.71
|
| CAS号 |
1467157-21-6
|
| 相关CAS号 |
1467829-71-5 (free acid);1467157-21-6 (sodium);
|
| PubChem CID |
89851851
|
| 外观&性状 |
White to light yellow solid
|
| tPSA |
141
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
19
|
| 分子复杂度/Complexity |
541
|
| 定义原子立体中心数目 |
2
|
| SMILES |
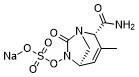
CC1=C[C@@H]2CN([C@@H]1C(=O)N)C(=O)N2OS(=O)(=O)[O-].[Na+]
|
| InChi Key |
WHHNOICWPZIYKI-IBTYICNHSA-M
|
| InChi Code |
InChI=1S/C8H11N3O6S.Na/c1-4-2-5-3-10(6(4)7(9)12)8(13)11(5)17-18(14,15)16/h2,5-6H,3H2,1H3,(H2,9,12)(H,14,15,16)/q+1/p-1/t5-,6+/m1./s1
|
| 化学名 |
sodium (2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl sulfate
|
| 别名 |
ETX2514 ETX-2514 ETX 2514 ETX2514 sodium Durlobactam sodium
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O: ~250 mg/mL (835 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3418 mL | 16.7090 mL | 33.4180 mL | |
| 5 mM | 0.6684 mL | 3.3418 mL | 6.6836 mL | |
| 10 mM | 0.3342 mL | 1.6709 mL | 3.3418 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 奈莫雷生盐酸盐
奈莫雷生盐酸盐
 E55888 HCL
E55888 HCL
 FITC-Dextran (MW 110000)
FITC-Dextran (MW 2000000)
FITC-Dextran (MW 110000)
FITC-Dextran (MW 2000000)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
