| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
NMDA receptor
|
|---|---|
| 体外研究 (In Vitro) |
由 G 蛋白激活的内向整流 K+ 通道 (GIRK)[1]。 通过电压箝法记录克隆NR1和NR2亚基rna表达异源NMDA受体的爪蟾卵母细胞,研究了非典型n -甲基- d -天冬氨酸(NMDA)受体拮抗剂Ifenprodil的作用。在-70 mV电压箝位的卵母细胞中,Ifenprodil对NR1A/NR2B受体的nmda诱导电流具有高亲和力抑制作用(IC50 = 0.34微米)。NR1A/NR2A受体对伊芬普罗地尔的亲和力(IC50 = 146 microM)比NR1A/NR2B受体低400倍。低浓度的伊芬普罗地尔作用于NR1A/NR2B受体时的抑制起效率明显慢于高浓度的伊芬普罗地尔作用于NR1A/NR2A受体时的抑制起效率。Ifenprodil/伊芬普罗地尔阻断NR1A/NR2B受体的开始和恢复不具有活性依赖性。低浓度的伊芬普罗地尔对NR1A/NR2B受体的抑制作用不依赖于电压。相反,高浓度伊芬普罗地尔对NR1A/NR2A受体的抑制作用部分依赖于电压,并且在超极化膜电位处比在去极化膜电位处对nmda诱导电流的抑制更大。在伊芬地尔的作用下,NMDA电流的逆转电位没有改变。伊芬普罗地尔可能作为NR1A/NR2A受体的弱开放通道阻滞剂。100微米的非芬普罗地尔对NR1A/NR2A受体的抑制程度不因细胞外甘氨酸浓度的变化而改变。然而,增加甘氨酸浓度可降低1 μ m非芬地尔对NR1A/NR2B受体的抑制作用。因此,伊芬普罗地尔作用于NR1A/NR2B受体的部分机制可能涉及对甘氨酸作用的非竞争性拮抗。这些结果表明,对爪蟾卵母细胞中表达的NR1A/NR2B和NR1A/NR2A受体,伊芬地尔的作用机制和拮抗剂的效力不同。[1]
G蛋白激活的内向纠偏K+通道(GIRK,也称为Kir3)受多种G蛋白偶联受体调节。GIRK通道的激活在降低大多数大脑区域的神经元兴奋性和心率方面起着重要作用。Ifenprodil是一种临床使用的脑血管扩张剂,可与几种受体相互作用,如α - 1肾上腺素能受体、n -甲基- d -天冬氨酸受体、血清素受体和西格玛受体。然而,各种临床相关作用的分子机制仍有待阐明。在这里,我们通过非洲爪蟾卵细胞表达测定来检测Ifenprodil/伊芬普罗地尔对GIRK通道的影响。在注射了GIRK1/GIRK2、GIRK2或GIRK1/GIRK4亚基mrna的卵母细胞中,伊芬prodil通过基础GIRK活性可逆地降低了内向电流。这种抑制作用是浓度依赖性的,但与电压和时间无关,这表明伊芬普罗地尔可能不是通道的开放通道阻滞剂。相反,其他Kir亚家族的Kir1.1和Kir2.1通道对伊芬普罗地尔不敏感。此外,克隆的kappa-阿片受体激活的GIRK电流反应同样被伊芬普罗地尔抑制。在细胞内施用伊芬普罗地尔时,没有观察到其抑制作用,也不受细胞外pH的影响,pH改变了未带电的质子化伊芬普罗地尔的比例,表明其作用于细胞外侧。在异丙地尔的存在下,乙醇诱导的GIRK电流也有所减弱。我们的研究结果表明,在亚微摩尔浓度或更高浓度下,伊芬地尔直接抑制GIRK通道可能有助于其一些治疗效果和不良副作用[2]。 在基于CPE的抗病毒试验中,Ifenprodil与nylidrin相当,PR8和HK的EC50值分别为6.6和16.9μM(表1)。盐酸克伦特罗仅对PR8具有强效活性,EC50值为9.4μM,而拉贝洛尔和依利地尔仅对HK具有微弱疗效,EC50值更高,高于44.0μM。相比之下,利多君、非诺特罗和班布特罗对任何病毒株都没有抗病毒活性。在进一步的实验中,我们测试了抑制化合物,甚至部分包括nylidrin、Ifenprodil、拉贝洛尔、依利普地尔和克伦特罗,以对抗其他甲型和乙型流感病毒(表2)。Nylidrin、Ifenprodil和克伦特罗对甲型H1N1流感病毒株一直有效,但它们的疗效在H3N2病毒株之间有所不同,在乙型流感病毒感染的细胞中没有抗病毒活性。正如预期的那样,剩下的两种化合物拉贝洛尔和依利普地尔对这些病毒株几乎没有影响,除了A/Brisbane/59/2007(H1N1),它对依利普迪尔部分敏感(EC50,53.1μM)。这些结果表明,nylidrin及其类似物Ifenprodil和克伦特罗在亚毒性浓度下可以可靠地抑制甲型H1N1流感病毒的感染[4]。 Ifenprodil与NMDA受体的相互作用降低了离子通道的开放状态,并抑制了Ca2+离子的流入。关于这一机制,依非普利具有神经保护、抗惊厥和镇痛作用。ifenprodil的结合位点最初被认为位于GluN2B亚基的氨基末端结构域(ATD)。后来,它可以在GluN1和GluN2B亚基之间的表面发现。根据第一个开发的配体,它被称为“ifenprodil结合位点”,可以与受体的不同其他结合位点相互作用。伊芬普利的NMDA亲和力非常高(IC50=13.3 nM,Ki=10 nM),但其选择性相当差[5]。 |
| 体内研究 (In Vivo) |
我们发现,无论是Ifenprodil还是布比卡因,都能剂量依赖性地产生运动功能和伤害感受的脊髓阻滞。在ED50的基础上,异环磷酰胺的效力为0.42(0.38-0.46)μmol;0.40(0.36-0.44)μmol)与布比卡因0.38(0.36-0.40)μmol相等(p>0.05);0.35(0.32-0.38)μmol)分别影响运动功能和伤害感受。在等麻醉剂量(ED25、ED50和ED75)下,在运动功能和伤害感受方面,Ifenprodil产生的持续时间大于布比卡因(差异p<0.05)。此外,伊芬普地尔和布比卡因的感觉阻滞持续时间均长于运动阻滞(差异p<0.05)。
结论:结果数据表明,在脊髓麻醉中,Ifenprodil具有剂量依赖性的局部麻醉作用。Ifenprodil显示出比运动阻滞更具感觉选择性的作用持续时间,而Ifenprodi的麻醉持续时间明显长于布比卡因。[3]
本研究的结果首次表明,鞘内注射Ifenprodil可引发运动功能和伤害性感觉的脊髓阻滞。Ifenprodil的脊髓麻醉效果与长效局部麻醉剂布比卡因相当。Ifenprodil和布比卡因显示出比运动阻滞更长的伤害性/感觉阻滞持续时间。在等麻醉剂量(ED25、ED50和ED75)下,Ifenprodil的脊髓麻醉持续时间大于。.. 我们的临床前数据显示,鞘内注射Ifenprodil在脊髓麻醉中产生了剂量依赖性的局部麻醉作用。Ifenprodil在脊髓麻醉中的效力与布比卡因相似,而Ifenprodil的脊髓麻醉持续时间大于布比卡因。此外,Ifenprodil和布比卡因表现出明显的感觉特异性过运动阻滞。Ifenprodil的神经阻滞值得进一步测试[3]。 体内代谢产物的鉴定[5] 对于体内代谢物,一只大鼠在腹腔注射20mgIfenprodil/kg大鼠后,分三个时期(0-8h、8-24h和24-48h)收集48小时的尿液。鉴定的代谢物如图7所示。仅在体内发现了三种代谢物(11、12和13)。代谢物11是邻羟基化、甲基化和葡萄糖醛酸化的结果。区域异构体葡糖苷酸12a和12b具有与8相同的质量(m/z 518),但显示出不同的裂解模式(见支持信息)。它们是从羟基化代谢物4开始产生的,这些代谢物还被葡萄糖醛酸化。与代谢物4类似的裂解模式允许识别相应的代谢物。此外,观察到两种具有额外O原子的代谢物13,但裂解模式无法明确指定O原子的位置。在加入PAPS后,在体外系统中鉴定出的硫酸盐10在大鼠尿液中没有检测到。然而,这种代谢物是否在样本尿液中或在储存过程中没有形成或分解仍有待阐明。 |
| 酶活实验 |
Microsomal incubations (phase I) (第一阶段)[5]
将Ifenprodil(235μg)溶解在含有氯化镁(100μL,45.5mM)的磷酸盐缓冲液(250μL,pH 7.4,0.1M)中。在加入150μL微粒体制剂(7mg蛋白质/mL)和5mg NADPH/H+后,将混合物在室温下在摇床中孵育。120分钟后,通过加入等体积的冷乙腈(-20°C)终止孵育。将混合物在冰浴中储存10分钟。随后,将样品在13000 rpm下离心8分钟,并在不进行进一步样品预处理的情况下通过HPLC-MS分析所得上清液。 Ifenprodil代谢降解率[5] 在校准研究中,将[strong>Ifenprodil(117.5μg)溶解在磷酸钠缓冲液(125μL,pH 7.4,0.1 M)中。加入氯化镁(50μL,45.5 mM)和大鼠肝微粒体悬浮液(100μL)。用100、80、60、40、20和0%的母体伊芬普利浓度(117.5μg)进行校准,用磷酸钠缓冲液稀释以获得所需浓度。120分钟后,通过加入冷CH3CN(500μL,-20°C)停止反应,并加入内标eliprodil(62.75μg,溶解在32.5μL CH3CN中)。将混合物在冰浴中储存10分钟,随后离心(13000rpm,8分钟)。用CH3CN/H2O(1/1)将上清液稀释1:40,并注入HPLC系统1。每个校准步骤进行三次。手动整合得到的TIC(m/z=326.2和m/z=348.15)。使用Microsoft Excel 2010计算eliprodil/Ifenprodil比值和所得校准曲线。 用溶解在磷酸钠缓冲液(125μL,pH 7.4,0.1 M)中的伊芬普利(117.5μg)进行代谢降解。加入氯化镁(50μL,45.5 mM)、大鼠肝微粒体(100μL)、NADPH(2.5 mg)和UDPGA(2 mg)。将该反应混合物摇动15、30、60、90和120分钟。然后,通过加入冷乙腈(500μL,-20°C)和依利脯啶(62.75μg,溶解在32.5μL CH3CN中)停止反应。将混合物在冰浴中储存10分钟,随后离心(13000rpm,8分钟)。用乙腈/H2O(1/1)将上清液稀释1:40,并注入HPLC系统1。每一步都进行了三次。由此产生的TIC(m/z 326.2和m/z 348.15)是手动集成的。使用Microsoft Excel 2010计算依普罗地尔/Ifenprodil比率和由此产生的依普罗地尔量。 |
| 细胞实验 |
为了检查细胞内Ifenprodil的作用,23 nl为10 mM ifenprodil或30 mM lidocaine N-ethyl bromide (QX-314) 溶解在蒸馏水中的mM利多卡因N-乙基溴(QX-314)通过使用Nanoliter注射器进行压力注射的额外移液管施用于卵母细胞,如前所述(Kobayashi等人,2003),然后连续记录卵母细胞电流约30-40 由于使用的爪蟾卵母细胞体积约为1 μl,细胞内Ifenprodil或QX-314的浓度被推测为∼225或∤674 μM。使用KaleidaGraph将数据拟合到标准逻辑斯谛方程中,以分析浓度-反应关系。EC50值,即产生该药物最大电流反应50%的药物浓度;IC25和IC50值,分别是将控制电流反应降低25%和50%的药物浓度;Hill系数(nH)由浓度-响应关系获得。[2]
化合物和细胞病变效应(CPE)减少试验[4] 使用了试验化合物盐酸尼利德林(~95%)、Ifenprodil(+)-酒石酸盐(≥98%)、盐酸拉贝洛尔(≥98%)、盐酸利托君(99.6%)、氢溴酸非诺特罗(≥98 0%)、依利普地尔(≥98%.)、盐酸克伦特罗(≥95%)和盐酸班布特罗(≥98%.)、M2抑制剂盐酸金刚烷胺(AMT;≥98%)和聚合酶抑制剂利巴韦林(RBV;≥98%.)、奥司他韦羧酸盐(OSV-C;≥98%。 免疫测定[4] 为了检测病毒蛋白,如前所述进行了蛋白质印迹分析。在nylidrin、OSV-C或RBV存在下,在35°C下用MOI为0.001的PR8病毒感染6孔板中生长至100%融合的MDCK细胞24小时。通过免疫印迹法检测病毒蛋白NP、HA和M1,分别检测相应的抗体:小鼠抗NP、兔抗HA2 和小鼠抗M1。第二抗体是辣根过氧化物酶(HRP)偶联的山羊抗小鼠或抗兔IgG。细胞β-肌动蛋白被用作负载对照。 菌斑检测[4] PR8感染的MDCK细胞被模拟处理或用单独的化合物处理。在35°C下24小时后,收集培养上清液以制备10倍的连续稀释液,用于感染接种在48孔板中的幼稚MDCK细胞1小时。PBS洗涤后,将其在33°C的覆盖培养基(含0.6%羧甲基纤维素和2µg/mL TPCK胰蛋白酶的MEM)中孵育三天。通过结晶紫染色计数斑块。 对于添加时间实验,在有或没有nylidrin的情况下,在4°C下用PR8(MOI,0.001)感染48孔板中的MDCK细胞1小时。用PBS洗涤以去除化学物质和未吸附的病毒后,在1、2和4小时p.i.(−)给药nylidrin。将表没食子儿茶素没食子酸酯用作阻断病毒进入的对照。随后,所有样品用PBS以5小时p.i.洗涤,然后在覆盖培养基下孵育三天,然后进行结晶紫染色。 共聚焦显微镜[4] 在荧光显微镜下,将PR8感染的MDCK细胞在有或没有nylidrin的情况下在37°C下孵育5小时。固定和渗透后,使用抗NP抗体和Alexa Fluor 488偶联的山羊抗小鼠IgG观察病毒NP;用4′,6-二脒基-2-苯基吲哚对核DNA进行复染。图像在蔡司LSM 700共聚焦显微镜上捕获,并使用ZEN软件进行分析。 为了研究病毒NP与细胞Rab5或Rab7的共定位,将A549细胞接种在4孔载玻片中(每孔4×104个细胞)。第二天,根据制造商的说明,使用脂质体2000用0.4μg pEGFP-Rab5或pEGFP-Rab 7转染细胞。24小时后,在4°C下,在100μM nylidrin存在或不存在的情况下,模拟感染或以10的MOI感染PR8 30分钟,然后在37°C下再感染4小时。使用上述相同的一抗,但用Alexa Fluor 633偶联的二抗探测病毒NP蛋白。 |
| 动物实验 |
For isolating liver microsomes and for in vivo metabolism studies of Ifenprodil, Wistar rats weighing 200–320 g from a local strain were used. [5]
In vivo metabolism studies of Ifenprodil [5] Ifenprodil was dissolved in 0.9% sterile saline solution. A single dose of 6.2 mg Ifenprodil tartrate was administered i.p. to one female rat (310 g, Wistar rat), which results in a dose of 20 mg/kg. A dose of 20 mg/kg ifenprodil was chosen, since traxoprodil, which has similar structure and pharmacological properties, was used in previous experiments in a comparable amount [12]. The rat was housed individually in a metabolism cage with water and powdered feed ad libitum. Urine was collected for 48 h in three intervals of 0–8 h, 8–24 h and 24–48 h after i.p. application of ifenprodil. The volumes of urine samples were recorded and the samples were stored at −20 °C until analysis or directly used. Extraction was carried out with RP-18 SPE cartridges, which were pre-conditioned with 1 mL H2O and 1 mL EtOH. After application of the urine samples, the cartridge was rinsed with 10 mL of H2O. Afterwards, Ifenprodil and the corresponding metabolites were eluted with 10 mL of EtOH. The solvent was removed under reduced pressure. The resulting solid was dissolved in CH3CN:H2O 1:1 and analyzed with the different HPLC systems. The aim of the study was to compare the proposed spinal anesthetic effect of Ifenprodil, an a1 adrenergic receptor antagonist, with that of the long-acting local anesthetic bupivacaine. Methods: After intrathecally injecting the rats with five different doses of each drug, the dose-response curves of Ifenprodil and bupivacaine were constructed to obtain the 50% effective dose (ED50). The spinal blockades of motor function and nociception of Ifenprodil were compared with that of bupivacaine.[3] Male Sprague–Dawley rats (300–350 g) were used. Intrathecal Ifenprodil caused significant motor or nociceptive blockade. Intrathecal Ifenprodil, as well as the long-lasting local anesthetic bupivacaine produced a dose-dependent local anesthetic effect in spinal anesthesia in rats (Fig. 1). The ED25s, ED50s, and ED75s of Ifenprodil and bupivacaine are presented in Table 1. On the ED50 basis, the potency of Ifenprodil in motor function and nociception was comparable to that of bupivacaine (Table 1, p > 0.05). [3] |
| 药代性质 (ADME/PK) |
NMDA受体拮抗剂伊芬普罗地尔(Ifenprodil)是开发新型GluN2B选择性NMDA受体拮抗剂的重要先导化合物。伊芬普罗地尔本身对GluN2B亚基具有高亲和力,但对NMDA受体的选择性较差。这一特性以及其快速的生物转化是伊芬普罗地尔的主要缺点。为了优化新型、更具选择性的GluN2B(NMDA)受体拮抗剂的开发,鉴定伊芬普罗地尔的主要代谢途径至关重要。本文通过LC-MS(n)实验生成并分析了伊芬普罗地尔的体外和体内I期及II期代谢产物。体外实验采用大鼠肝微粒体和多种辅助因子生成I期和II期代谢产物。将伊芬普罗地尔应用于大鼠并分析其尿液,从而鉴定出多种体内代谢产物。酚类化合物是伊芬普罗地尔代谢最不稳定的结构单元,因为葡萄糖醛酸苷7和8是其主要代谢产物。[5] 据报道,伊芬普罗地尔的生物利用度较低。在人体中,其血浆峰浓度出现在给药后约30分钟。[9] 然而,伊芬普罗地尔的生物转化及其代谢产物的结构尚未被阐明。因此,鉴定伊芬普罗地尔代谢不稳定的位点可能有助于开发与伊芬普罗地尔结合位点相互作用的代谢更稳定的药物。本文报道了我们对伊芬普罗地尔体外和体内I期和II期代谢的研究。 [5]
伊芬普罗地尔的碎片化 [5] 伊芬普罗地尔的MSn实验揭示了一种碎片化模式,该模式可被认为是伊芬普罗地尔结构的典型特征(图1)。第一步碎片化是仲醇的脱水(m/z 308.2003)。进一步的碎片化导致m/z 293.1752的碎片,这可解释为甲基的丢失。伊芬普罗地尔的另一个重要碎片化是C-N(哌啶)单键的断裂,导致形成m/z 176.1424的苄基哌啶部分。三苯基正离子(m/z 91.0511)的释放进一步证实了该碎片的存在。母体化合物伊芬普罗地尔的这种碎片化模式是解释所生成代谢物碎片化研究的基础,并提供了有关所形成代谢物结构的信息。 体外I期代谢物的鉴定和碎片化[5] I期转化产生了多种代谢物,包括两种N-脱烷基化产物和几种M+O代谢物(图2,补充信息)。N-脱烷基化代谢物被鉴定为4-苄基哌啶(1)和氧化的4-苄基哌啶-2-酮(2)。哌啶代谢物1和2的结构通过碎片化实验确定。两种代谢物的主要碎片均为三苯基正离子,其精确质量分别为 m/z 91.0510 和 m/z 91.0518(见补充信息)。此外,还鉴定出六种 I 期代谢物,其 m/z 为 342 [M+O+H]+,这是由于引入了一个额外的氧原子(图 3a)。这些代谢物通过 MSn 实验进行分析。分析结果表明,氧化发生在哌啶环 (3)、苯基部分的邻位、间位和对位 (4a–4c)、酚羟基的邻位 (5) 以及形成 N-氧化物的氮原子 (6,图 3b)。三种代谢物 4a–4c 显示出相同的碎片模式,表明它们的结构非常相似。单羟基化代谢物 3 的羟基被归属于哌啶杂环。第一个线索是碎片 m/z 192。该质量比母体化合物相应碎片的质量高 16 amu。随后的碎片化产生了 m/z 174 的碎片,代表脱水的苄基哌啶。这些碎片证实了羟基的位置,因为只有当羟基位于哌啶部分时,才会出现碎片 m/z 174(图 4)。N-氧化物 6 由碎片 m/z 192.1358(氧化的苄基哌啶)和 m/z 174 [苄基哌啶 + O + H – H₂O]⁺ 鉴定。因此,额外的氧原子只能位于伊芬普罗地尔的苄基哌啶部分。与伊芬普罗地尔(9.4 分钟)相比,保留时间略有增加(10.8 分钟),表明存在 N-氧化物。由于N-氧化物的亲脂性略有增加,其保留时间通常比母体化合物更长。 体外发现的II相代谢物[5] 在II相反应中,伊芬普罗地尔(Ifenprodil)理论上可以转化为葡萄糖醛酸苷、硫酸盐和甲基化儿茶酚衍生物,如曲索普罗地尔(Traxoprodil)所述。β-葡萄糖醛酸苷和硫酸盐可由伊芬普罗地尔中存在的羟基直接反应生成。此外,I相反应的代谢物可在II相反应中发生结合。甲基化儿茶酚衍生物只有在苯酚邻位羟基化生成儿茶酚衍生物后才能生成(参见代谢物5)。 在体外,通过向微粒体孵育混合物中添加UDPGA(不添加NADPH/H+)来研究葡萄糖醛酸化反应。在正负离子模式下均记录了葡萄糖醛酸苷 7 [M+Glu+H]+ 的观测信号。正离子模式下,β-葡萄糖醛酸苷代谢物 7 的碎片化显示出两条主要的碎片化途径。与伊芬普罗地尔及其衍生物类似,观察到脱水(正离子模式下 m/z 484.2346)。另一方面,葡萄糖醛酸被裂解,产生碎片 m/z 326.2134 ([伊芬普罗地尔+H]+)。这两种碎片化同时发生,并最终产生相同的碎片 m/z 308.2040(参见 SI)。在负离子模式下也观察到了类似的碎片。此外,在负离子模式下还鉴定出了葡萄糖醛酸的碎片。碎片 m/z 193 代表葡萄糖醛酸根阴离子,而 m/z 175 对应于脱水葡萄糖醛酸。随后失去水和 CO2 生成碎片 m/z 113,这是葡萄糖醛酸苷的特征(图 5)。 将伊芬普罗地尔与 NADPH/H+ 和 UDPGA 孵育后,生成了额外的葡萄糖醛酸苷 8(图 2),这是在 I 期生物转化过程中产生的儿茶酚 5 发生葡萄糖醛酸化后形成的。同样的儿茶酚 5 也可以被儿茶酚 O-甲基转移酶甲基化。向孵育混合物中加入S-腺苷甲硫氨酸(SAM)和NADPH/H+后,生成了m/z为356.1965的化合物,该化合物的质量与甲基化儿茶酚9的质量相符。化合物9的碎片化(图6)与伊芬普罗地尔的碎片化(见图1)类似。未取代的苄基哌啶碎片m/z 176.1444的生成有力地证明了甲氧基化发生在酚羟基上,而非苄基上。此外,m/z 338.2129、163 和 137 碎片的质量比相应的伊芬普罗地尔碎片的质量高 30 amu。 体内代谢物的鉴定[5] 对于体内代谢物,在腹腔注射 20 mg/kg 伊芬普罗地尔后,收集一只大鼠 48 小时内的尿液,分为三个时间段(0-8 小时、8-24 小时和 24-48 小时)。鉴定出的代谢物如图 7 所示。三种代谢物(11、12 和 13)仅在体内发现。代谢物 11 是邻位羟基化、甲基化和葡萄糖醛酸化的产物。区域异构体葡萄糖醛酸苷 12a 和 12b 与化合物 8 具有相同的质量 (m/z 518),但显示出不同的碎片模式(参见补充信息)。它们由羟基化代谢物 4 进一步葡萄糖醛酸化生成。与代谢物 4 相似的碎片模式使得相应的代谢物得以鉴定。此外,还观察到两种含有额外氧原子的代谢物 13,但其碎片模式无法明确确定氧原子的位置。在体外系统中添加 PAPS 后鉴定出的硫酸盐 10 未在大鼠尿液中检测到。然而,该代谢物是否未生成,或在尿液样本中或储存过程中分解,仍有待阐明。 伊芬普罗地尔在最初阶段(0-8 小时)迅速排泄。在此期间,伊芬普罗地尔是主要化合物之一,与葡萄糖醛酸苷 7 和 11 一起,假设所有化合物的电离因子相当。在最初两个时间段(0-8 小时,8-24 小时)内,葡萄糖醛酸化被观察到是主要的代谢途径。即使在尿液收集的最后一个时间段(24-48 小时)仍检测到伊芬普罗地尔,表明即使在 24 小时后,伊芬普罗地尔仍以未修饰的形式排出体外(图 8)。 伊芬普罗地尔的代谢稳定性 [5] 在体外实验中,测定了伊芬普罗地尔在大鼠肝微粒体、NADPH/H+ 和 UDPGA 存在下的代谢稳定性。选择这些辅因子是为了生成体内观察到的主要代谢物。为了精确定量伊芬普罗地尔,首先记录了校准曲线。将不同用量的伊芬普罗地尔(相当于微粒体孵育用量的20%、40%、60%、80%和100%)与不含NADPH/H+的反应混合物进行孵育。此外,还添加了依利普罗地尔作为内标(IS)。以伊芬普罗地尔/IS的比值对所用伊芬普罗地尔的用量作图,结果得到良好的回归系数(见补充信息)。在长达120分钟的六个孵育周期后,测定代谢活性混合物中伊芬普罗地尔的浓度。 出乎意料的是,与NADPH/H+和UDPGA一起孵育60分钟后,伊芬普罗地尔的浓度反而增加。这一结果可以用葡萄糖醛酸苷7的快速分解来解释,这导致伊芬普罗地尔的再生。该理论通过仅用 NADPH/H+ 孵育伊芬普罗地尔而不添加 UDPGA 得到证实,结果显示伊芬普罗地尔的含量持续下降(图 9)。在两种辅因子共同孵育 60 分钟后,仍有 86 ± 2% 的伊芬普罗地尔未被检测到。仅生成 I 期代谢物后,60 分钟后伊芬普罗地尔的含量降至 92.8 ± 2%。因此,伊芬普罗地尔的主要生物转化是由葡萄糖醛酸化反应引起的。这一观察结果与体内实验结果相符,体内实验也发现葡萄糖醛酸苷 7 和 8 是主要代谢物。 伊芬普罗地尔是开发高效选择性 GluN2B 选择性 NMDA 受体拮抗剂的重要先导化合物,本文对其生物转化进行了分析。在体外实验中,N-去烷基化、两个芳香环的羟基化以及哌啶部分的羟基化被确定为可能的反应。在II期实验中,观察到了酚的葡萄糖醛酸化。此外,在与相应的辅因子SAM和PAPS孵育后,检测到了甲氧基化和硫酸化代谢物。对大鼠尿液样本的分析鉴定出了葡萄糖醛酸苷、羟基化和甲氧基化代谢物。葡萄糖醛酸苷7被鉴定为主要代谢物。 总之,伊芬普罗地尔的酚是易发生生物转化的主要结构单元。特别是,羟基的葡萄糖醛酸化被确定为体外和体内的主要代谢途径。这些结果清楚地表明,为了获得代谢更稳定的GluN2B选择性NMDA受体拮抗剂,应进行生物等排替换以取代酚。[5] |
| 毒性/毒理 (Toxicokinetics/TK) |
小鼠口服LD50为320 mg/kg。行为学:睡眠时间改变(包括翻正反射改变);行为学:运动活性改变(特异性检测)。Arzneimittel-Forschung. Drug Research., 21(1992), 1971 [PMID:4400568]
伊芬普罗地尔与中枢神经系统其他受体(α1、5-HT、σ1、σ2受体)的相互作用会导致不良副作用,例如运动功能障碍和血压降低。尽管如此,伊芬普罗地尔仍是合理设计新型GluN2B选择性拮抗剂的重要先导化合物,这些拮抗剂有望成为治疗危及生命的中枢神经系统疾病的药物[5]。 |
| 参考文献 |
|
| 其他信息 |
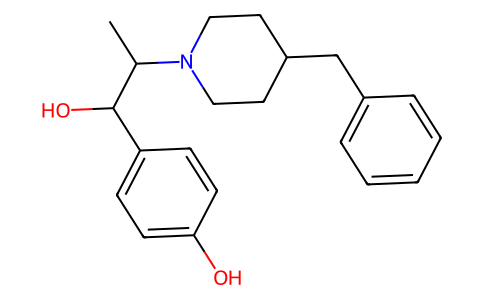
4-[1-羟基-2-[4-(苯甲基)-1-哌啶基]丙基]苯酚是哌啶类化合物。
N-甲基-D-天冬氨酸(NMDA)受体(NMDARs)是离子型谷氨酸受体家族的成员,在脑发育和神经功能中发挥着关键作用。NMDARs是异源四聚体,通常由GluN1和GluN2A-D亚基的二聚体组成,每个亚基本身由N端结构域(NTD)、配体结合结构域(LBD)、跨膜结构域和C端胞质结构域组成。激动剂甘氨酸(或D-丝氨酸)与GluN1亚基的LBD结合,谷氨酸与GluN2亚基的LBD结合,是通道激活的调节机制。此外,已知变构调节剂能够结合NMDAR的N端结构域(NTD),形成另一层调控机制。伊芬普罗地尔就是这样一种变构调节剂,它最早于20世纪90年代被发现能够结合NMDAR,特别是那些含有GluN2B亚基的NMDAR。进一步的研究表明,伊芬普罗地尔能够牢固地结合在相邻的GluN1和GluN2B NTD的亚基间界面,并在此发挥非竞争性拮抗剂的作用。尽管伊芬普罗地尔因其在精神疾病(包括成瘾和抑郁症)中的潜在神经调节活性而备受关注,但研究也表明它具有免疫调节作用。在一项针对能够减少由H5N1流感病毒感染引起的细胞死亡的化合物的无偏筛选中,发现伊芬普罗地尔对H5N1引起的肺损伤具有保护作用,部分原因是其能够减轻H5N1引起的细胞因子风暴,并减少中性粒细胞、自然杀伤细胞和T细胞在肺部的浸润。目前正在进行一项IIb/III期临床试验(NCT04382924),以研究伊芬普罗地尔治疗COVID-19的潜在疗效。 伊芬普罗地尔是一种口服生物利用度高的N-甲基-D-天冬氨酸(NMDA)受体拮抗剂,具有潜在的中枢神经系统(CNS)刺激、神经保护、抗炎和抗纤维化活性。伊芬普罗地尔给药后,靶向并结合谷氨酸能NMDA受体(NMDARs),特别是甘氨酸结合NMDA受体亚基1(GluN1)和亚基2(谷氨酸结合NMDA受体亚基2;NMDA型亚基2B;GluN2B),从而抑制NMDAR信号传导。这可抑制神经元兴奋性毒性,并可能增强认知功能。此外,伊芬普罗地尔还抑制G蛋白偶联内向整流钾(GIRK)通道,并与α1肾上腺素能受体、5-羟色胺受体相互作用,激活σ受体。伊芬普罗地尔通过其对NMDA受体以及可能的σ-1受体的作用发挥抗炎作用。尽管确切机制尚未完全阐明,但该药物可减少中性粒细胞和T细胞向肺部的浸润,并阻止促炎细胞因子的释放。这可能导致肺部炎症反应减轻,抑制肺纤维化,并可能减轻咳嗽的严重程度。NMDA受体是由四个亚基组成的多聚体离子型谷氨酸受体,表达于多种细胞和器官,例如脑、肺以及T细胞和中性粒细胞。 伊芬普罗地尔抑制GIRK通道的特征[2] 本研究表明,伊芬普罗地尔以独特的方式在纳摩尔或更高浓度下抑制脑型GIRK1/2和GIRK2通道以及心脏型GIRK1/4通道。伊芬普罗地尔对GIRK通道的抑制作用呈浓度依赖性,但与电压和时间无关,主要影响瞬时电流,并在每个电压脉冲期间保持稳定的抑制百分比。我们的结果还表明,伊芬普罗地尔是从细胞膜外侧作用于通道。另一方面,胞外Ba²⁺和Cs⁺(典型的Kir通道阻滞剂,它们通过阻塞开放通道的孔道发挥作用)的阻滞作用则表现出浓度依赖性、强烈的电压依赖性和时间依赖性,对瞬时电流的影响相对较小,但在电压脉冲结束时对稳态电流有显著抑制作用(Lesage等,1995)。这些观察结果表明,伊芬普罗地尔可能引起GIRK通道的构象变化,但并不像Ba²⁺和Cs⁺那样作为开放通道阻滞剂发挥作用。伊芬普罗地尔对GIRK电流的阻断不完全,其作用机制可能也与此有关。在本研究中,伊芬普罗地尔同样抑制了卵母细胞中基础游离G蛋白βγ亚基、κ阿片受体激活介导的G蛋白或乙醇诱导的GIRK电流。进一步的单通道实验研究可能有助于理解伊芬普罗地尔对GIRK通道的作用机制。 此外,伊芬普罗地尔对GIRK1/4通道的抑制效力高于GIRK1/2和GIRK2通道。尽管在所测试的最高浓度下,伊芬普罗地尔对不同通道的抑制效力顺序为GIRK2>GIRK1/2≥GIRK1/4通道,但差异无统计学意义。此外,其他Kir通道亚家族中的Kir1.1和Kir2.1通道对伊芬普罗地尔不敏感。利用GIRK/Kir1.1和GIRK/Kir2.1嵌合通道以及GIRK突变通道的进一步研究,或许能够阐明伊芬普罗地尔对GIRK通道作用的关键位点。此外,GIRK通道的高分辨率结构分析可能有助于表征其结合位点。另外,尽管氟哌啶醇在结构上与伊芬普罗地尔相关(Williams,2001),但氟哌啶醇对GIRK1/2和GIRK1/4通道的抑制作用较弱,且作用方式类似(Kobayashi等,2000)。这些药物对GIRK通道的不同作用可能源于它们不同的化学结构,也可能源于它们在GIRK通道上的不同结合位点。对GIRK通道结构与伊芬普罗地尔结构之间关系的研究可能为设计高效GIRK抑制剂候选药物提供基础。 临床和药理学意义[2] 据报道,单次服用临床剂量后,人体血浆中伊芬普罗地尔的浓度约为0.1 μM(Aventis Pharma公司数据)。在动物实验中,肌注放射性标记的伊芬普罗地尔后,脑和心脏中的浓度分别比血液中的浓度高约5-8倍和5-10倍(Nakagawa等,1975)。因此,本研究结果表明,在临床相关浓度下,伊芬普罗地尔可能抑制脑和心脏中的GIRK通道。在生理条件下,GIRK通道的激活会诱导K+外流,导致膜超极化(North,1989);而GIRK通道的抑制则会导致膜电位去极化,从而增加细胞兴奋性(Kuzhikandathil和Oxford,2002)。因此,在临床应用中,伊芬普罗地尔可能通过抑制广泛表达于神经系统和心房的GIRK通道,影响多种脑和心脏功能(Kobayashi等,1995;Karschin等,1996)。 GIRK2基因敲除小鼠表现出自发性癫痫发作,并且比野生型小鼠更容易被戊四唑(一种GABAA受体拮抗剂)诱发癫痫发作(Signorini等,1997)。此外,与野生型小鼠相比,GIRK基因敲除小鼠神经元的静息膜电位发生去极化(Lüscher等,1997;Torrecilla等,2002)。高剂量的伊芬普罗地尔可增强某些致惊厥药物(包括戊四唑)诱发的癫痫发作(Mizusawa等,1976),尽管伊芬普罗地尔已被证明具有抗惊厥作用(Thurgur和Church,1998;Yourick等,1999),这可能是由于其抑制了NMDA受体通道(Williams,2001)和Ca2+通道(Church等,1994;Bath等,1996)。尽管伊芬普罗地尔具有抗惊厥作用,但其对神经元GIRK通道的强效阻断可能通过增加神经元兴奋性而导致癫痫易感性增加。 有趣的是,GIRK2基因敲除小鼠在三种焦虑测试(高架十字迷宫、明暗箱和树冠测试)中均表现出焦虑减轻和运动活性增加(Blednov等,2001)。伊芬普罗地尔在高架十字迷宫测试中对MF1小鼠具有抗焦虑作用,可增加其运动量(Fraser等,1996),但在明暗箱探索测试中未观察到抗焦虑作用,且对Wistar大鼠的运动活性无影响(Mikolajczak等,2003)。这种差异可能是由行为测试的不同造成的,包括实验装置中两个明暗隔间的比例不同和/或动物种类不同。一项临床研究表明,伊芬普罗地尔可以改善脑血管疾病后遗症患者的焦虑、自发性降低和忧郁症状(Otomo等,1976)。因此,伊芬普罗地尔对神经元GIRK通道的抑制作用可能部分解释了其对焦虑和活动减少的临床疗效,这些疗效也见于某些神经精神疾病。在心脏中,乙酰胆碱通过激活M2毒蕈碱型乙酰胆碱受体来打开心房GIRK通道,最终导致心率减慢(Brown和Birnbaumer,1990)。伊芬普罗地尔治疗期间,除降压作用外,还观察到窦性心动过速(Carron等,1971;Young等,1983;Yajima等,1987)。伊芬普罗地尔对毒蕈碱型乙酰胆碱受体无显著亲和力(Chenard等,1991)。本研究表明,亚微摩尔浓度或更高浓度的伊芬普罗地尔可抑制心脏型GIRK1/4通道,这些通道大量存在于心房中(Krapivinsky等,1995)。因此,在临床实践中,伊芬普罗地尔也可能抑制心房GIRK通道。GIRK1或GIRK4基因敲除小鼠表现出轻度心动过速(Bettahi等,2002)。此外,伊芬普罗地尔的降压作用可能诱导交感神经系统的代偿性激活,而交感神经系统在心率的刺激调节中起着重要作用。综上所述,我们的数据表明,伊芬普罗地尔治疗期间的窦性心动过速可能部分与心房GIRK通道的抑制有关。 伊芬普罗地尔影响动物体内与乙醇相关的行为改变,例如抑制遗忘效应和包括惊厥在内的戒断症状(Malinowska等,1999;Napiórkowska-Pawlak等,2000;Narita等,2000)。乙醇激活GIRK通道(Kobayashi等,1999;Lewohl等,1999)。本研究表明,伊芬普罗地尔抑制了乙醇诱导的GIRK1/2电流。有趣的是,GIRK2基因敲除小鼠表现出乙醇诱导的条件性味觉厌恶和条件性位置偏好降低(Hill等,2003),并且与野生型小鼠相比,它们对一些急性乙醇效应的敏感性较低,包括抗焦虑作用、习惯性运动兴奋和急性乙醇给药后操作诱发的惊厥(Blednov等,2001)。综上所述,伊芬普罗地尔可能抑制GIRK相关的乙醇效应。 吗啡是一种常用的强效镇痛药,它优先与μ-阿片受体结合,并发挥多种药理作用,包括镇痛、欣快和依赖性(Gutstein和Akil,2001)。 μ-阿片受体与G蛋白介导的信号转导通路偶联,该通路涉及GIRK通道、腺苷酸环化酶、Ca2+通道和磷脂酶C(Ikeda等,2002)。虽然吗啡在动物中产生条件性位置偏好,表明其具有奖赏效应,但伊芬普罗地尔预处理会抑制吗啡产生的奖赏效应(Suzuki等,1999)。然而,伊芬普罗地尔对阿片受体没有显著的亲和力(Chenard等,1991)。本研究表明,伊芬普罗地尔抑制了G蛋白介导的GIRK电流。确定GIRK通道功能是否参与吗啡的奖赏效应可能很重要。有趣的是,GIRK基因敲除小鼠表现出可卡因自我给药减少(Morgan等,2003)。在一项临床报告中,地昔帕明(一种GIRK通道和去甲肾上腺素转运体抑制剂)(Kobayashi等,2004b)促进了可卡因的初始戒断(Gawin等,1989)。因此,选择性GIRK抑制剂可能成为治疗可卡因滥用者的潜在药物。进一步研究伊芬普罗地尔对GIRK基因敲除小鼠的影响,或许能够阐明伊芬普罗地尔通过GIRK介导的作用在吗啡和可卡因成瘾中的作用机制。[2] 多核体形成实验证实,尼利地林通过阻断HA的pH依赖性构象变化,靶向HA2介导的膜融合。如上所述,抗病毒疗效对病毒亚型的依赖性也为这一假设提供了进一步的证据:尼利德林、伊芬普罗地尔和克仑特罗对所有测试的A/H1N1毒株均有效;相比之下,在测试的H3N2毒株中,尼利德林和伊芬普罗地尔仅对A/Hong Kong/8/1968和A/Seoul/11/1988毒株有有限的疗效(表1和表2)。这一观察结果令人关注,因为大多数HA2融合抑制剂,无论是治疗性抗体还是小分子药物,其抗病毒效力往往取决于病毒亚型或HA组;I组包括H1、H2和H5,而II组包括H3和H7。这些化合物对H3N2亚型病毒的毒株特异性活性表明,通过化学修饰优化nylidrin可能是鉴定广谱融合抑制剂的一种可行方法。[4]甲型流感病毒是主要的人类呼吸道病原体之一,每年都会引发季节性流行和难以预测的周期性大流行。尽管疫苗和抗病毒药物已在临床上得到应用,但该病毒的抗原多样性和耐药性使其对公共卫生构成持续威胁,凸显了开发新型抗病毒药物的必要性。在一项基于细胞培养的高通量筛选中,β2-肾上腺素能受体激动剂nylidrin被鉴定为一种抗甲型流感病毒的化合物。该分子对多种H1N1亚型分离株有效,但对H3N2亚型的活性有限,且活性取决于毒株。通过研究其化学类似物的抗病毒活性,我们发现伊芬普罗地尔和克仑特罗对甲型H1N1流感病毒株也具有可靠的抑制作用。基于场的药效团模型结合活性化合物和非活性化合物的比较,揭示了苯氨基乙醇衍生物正负静电模式的重要性。加入时间实验和核蛋白NP细胞内定位的可视化结果表明,尼利地尔抑制了病毒生命周期的早期步骤。最终,我们发现尼利地尔通过阻断HA在酸性pH条件下的构象变化,靶向血凝素2(HA2)介导的膜融合。在小鼠模型中,用尼利地尔预孵育小鼠适应性甲型H1N1流感病毒可完全阻断鼻内病毒感染。本研究表明,尼利德林可为开发直接作用于甲型流感病毒入侵的抑制剂提供核心化学骨架。[4] |
| 分子式 |
C21H27NO2
|
|---|---|
| 分子量 |
325.44458
|
| 精确质量 |
475.221
|
| 元素分析 |
C, 77.50; H, 8.36; N, 4.30; O, 9.83
|
| CAS号 |
23210-56-2
|
| 相关CAS号 |
23210-58-4 (tartrate);23210-56-2;66157-43-5 (tartrate);
|
| PubChem CID |
3689
|
| 外观&性状 |
Typically exists as White to off-white solid at room temperature
|
| 密度 |
1.14 g/cm3
|
| 沸点 |
493.5ºC at 760 mmHg
|
| 熔点 |
148ºC
|
| 闪点 |
248.7ºC
|
| 蒸汽压 |
1.48E-10mmHg at 25°C
|
| LogP |
1.584
|
| tPSA |
158.76
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
24
|
| 分子复杂度/Complexity |
353
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC(C(C1=CC=C(C=C1)O)O)N2CCC(CC2)CC3=CC=CC=C3
|
| InChi Key |
UYNVMODNBIQBMV-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H27NO2/c1-16(21(24)19-7-9-20(23)10-8-19)22-13-11-18(12-14-22)15-17-5-3-2-4-6-17/h2-10,16,18,21,23-24H,11-15H2,1H3
|
| 化学名 |
4-[2-(4-benzylpiperidin-1-yl)-1-hydroxypropyl]phenol
|
| 别名 |
Ifenprodil; 23210-56-2; Dilvax; Vadilex; Creocral; Ifenprodilum; 4-(2-(4-benzylpiperidin-1-yl)-1-hydroxypropyl)phenol; 4-[2-(4-benzylpiperidin-1-yl)-1-hydroxypropyl]phenol;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0728 mL | 15.3638 mL | 30.7276 mL | |
| 5 mM | 0.6146 mL | 3.0728 mL | 6.1455 mL | |
| 10 mM | 0.3073 mL | 1.5364 mL | 3.0728 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
