| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
描述:普利诺司他(曾用名AG3340和KBR-9896)是一种合成的羟肟酸类似物,是一种新型、广谱、强效的口服生物活性金属蛋白酶(MMP)抑制剂,对MMP-1、MMP-3和MMP-9的IC50值分别为79、6.3和5.0 nM。它具有潜在的抗肿瘤活性。普利诺司他可抑制基质金属蛋白酶(MMP)(特别是MMP-2、9、13和14),从而诱导细胞外基质降解,并抑制血管生成、肿瘤生长、侵袭和转移。作为一种亲脂性药物,普利诺司他可穿过血脑屏障。
| 靶点 |
MMP-9 (IC50 = 5 nM); MMP-9 (IC50 = 0.26 nM); MMP-2 (Ki = 0.05 nM); MMP-1 (IC50 = 79 nM); MMP-3 (IC50 = 6.3 nM); MMP-3 (Ki = 0.3 nM); collagenases 3 (Ki = 0.03 nM)
Prinomastat (AG3340) is a synthetic, broad-spectrum matrix metalloproteinase inhibitor. It inhibits the proteolytic activity of MMPs, including the caseinolytic activity of MMP-3 (stromelysin-1) and the gelatinolytic activity of other MMPs [2]. |
|---|---|
| 体外研究 (In Vitro) |
普利诺司他(AG3340;0.1–1 µg/mL;4 天;C57MG/Wnt1 细胞)抑制 Wnt1 诱导的 MMP-3 的产生。普利诺司他可逆转 Wnt1 诱导的 EMT 和 β-catenin 转录活性 [1]。当 L/Wnt3a 细胞与 CT7 细胞共培养时,CT7 细胞中的 Topflash 活性增加;类似地,将 L/Wnt3a 细胞和过表达 MMP-3 的 C57MG 细胞与 CT7 细胞共培养,可使 CT7 细胞中的 Topflash 荧光素酶活性超过观察到的水平。普利诺司他(AG3340)抑制这些效应。Erk1/2 和细胞周期蛋白 D1 磷酸化表达的降低与普利诺司他抑制 C57MG/Wnt1 细胞进入 S 期相关。随后,我们利用体外划痕实验研究了普利诺司他(Prinomastat)对Wnt1诱导的细胞迁移的影响。正如预期的那样,C57MG/Wnt1细胞的迁移能力是C57MG细胞的1.8倍。 Prinomastat 可拮抗 Wnt1 对 C57MG/Wnt1 细胞中波形蛋白细胞分布的影响 [1]。
抑制 MMP 活性:在明胶酶谱分析中,将凝胶与 AG3340 (20 μg/ml) 孵育过夜,可完全抑制 C57MG/Wnt1 细胞条件培养基中 MMP 的明胶酶活性,证实观察到的条带代表 MMP 活性 [2]。 逆转 Wnt 诱导的 EMT:用 AG3340 (10 μg/ml) 处理 C57MG/Wnt1 细胞或 Wnt3a 处理的 C57MG 细胞,可逆转上皮-间质转化表型,使细胞恢复上皮形态。这与β-catenin从细胞核重新定位到细胞膜有关[2]。 抑制β-catenin转录活性:AG3340(10 μg/ml)显著抑制C57MG/Wnt1细胞和Wnt3a处理的C57MG细胞中β-catenin的转录活性,该结果通过Topflash荧光素酶报告基因检测法测定[2]。 抑制细胞增殖:用AG3340(10 μg/ml,处理24-48小时)处理可抑制Wnt1诱导的细胞增殖。FACS分析显示,AG3340使C57MG/Wnt1细胞S期比例从17.6%降低至8.4%。这伴随着细胞周期蛋白D1表达和Erk1/2磷酸化的降低[2]。 抑制细胞迁移:在伤口愈合实验中,AG3340(10 μg/ml,24小时)显著抑制C57MG/Wnt1细胞的迁移,使迁移细胞数量减少至与未转染的C57MG细胞相当的水平[2]。 细胞骨架重组:用AG3340(10 μg/ml)处理C57MG/Wnt1细胞可逆转Wnt1诱导的波形蛋白从细胞突起(与肌动蛋白丝相关)的重组,使其恢复到核周分布[2]。 下调MMP-3表达:用AG3340(10-50 μg/ml,4天)孵育C57MG/Wnt1细胞可显著下调MMP-3表达。如酪蛋白酶谱和蛋白质印迹实验所示,MMP-3 的产生和分泌减少。这与 MMP-3 启动子活性降低有关,表明 MMP 抑制剂会下调 Wnt 诱导的 MMP-3 转录 [2]。 |
| 体内研究 (In Vivo) |
在人纤维肉瘤小鼠模型(HT1080)中,从肿瘤接种后第3天开始,至第6天结束,每天腹腔注射50 mg/kg的普利司他(Primastat),持续14-16天。小鼠对普利司他反应良好,未出现体重减轻或任何副作用。普利司他的半衰期仅为1.6小时,且具有良好的肿瘤生长抑制作用[1]。研究人员在多种肿瘤模型中研究了AG3340,这是一种强效的金属蛋白酶(MMP)抑制剂,对明胶酶(MMP-2和-9)、MT-MMP-1(MMP-14)和胶原酶-3(MMP-13)具有皮摩尔级的亲和力。AG3340在小鼠体内表现出剂量依赖性的药代动力学特征,腹腔注射和口服给药后均耐受良好。在人类肿瘤模型中,无论在肿瘤植入后早期还是晚期开始给药,AG3340 均能显著延缓肿瘤生长,但并非所有已建立的肿瘤类型均对 AG3340 有反应。研究人员在三种模型中探讨了剂量-反应关系:COLO-320DM 结肠癌模型、MV522 肺癌模型和 MDA-MB-435 乳腺癌模型。在结肠癌和肺癌模型中观察到剂量依赖性的肿瘤生长抑制(每日两次,每次 12.5-200 mg/kg);在第三种模型(乳腺癌)中,最低测试剂量(50 mg/kg,每日两次)的 AG3340 即可产生最大抑制作用。在另一种模型中,AG3340(100 mg/kg,每日一次,腹腔注射)显著抑制了 U87 胶质瘤的生长并延长了动物的生存期。AG3340 还抑制了携带雄激素非依赖性 PC-3 前列腺肿瘤的裸鼠的肿瘤生长并延长了其生存期。在第六个模型(KKLS胃癌模型)中,AG3340并未抑制肿瘤生长,但增强了紫杉醇的疗效。重要的是,AG3340显著降低了肿瘤血管生成(通过CD-31染色评估)和细胞增殖(通过溴脱氧尿苷掺入评估),并增加了肿瘤坏死和凋亡(通过苏木精-伊红染色和TUNEL染色评估)。这些作用具有模型依赖性,但血管生成普遍受到抑制。在MV522肺癌模型中,AG3340的治疗指数优于细胞毒性药物卡铂和紫杉醇。联合用药时,AG3340增强了这些细胞毒性药物的疗效,且不影响药物耐受性。此外,在静脉转移模型中,当与卡铂或紫杉醇联合使用时,AG3340可减少小鼠黑色素瘤(B16-F10)肺部病灶的数量。这些研究直接支持在正在进行的肺癌和前列腺癌晚期恶性肿瘤患者的一线联合化疗中使用AG3340。[3]
普利诺司他治疗组和对照组的平均增生性玻璃体视网膜病变(PVR)评分分别为2.62和3.57(p = 0.038;Wilcoxon秩和检验)。对照组中76%的兔子出现具有临床意义的PVR伴视网膜脱离(PVR≥3级),而普利诺司他治疗组兔子的这一比例为51%。结论:在利用分散酶诱导增殖性玻璃体视网膜病变(PVR)的实验模型中,玻璃体内注射普利诺司他可降低PVR的发生率[4]。 转基因小鼠乳腺肿瘤发生延迟:在一项预防性治疗研究中,雌性MMTV-Wnt1转基因小鼠(可自发发生乳腺肿瘤)从6周龄开始,每周两次腹腔注射AG3340(50 mg/kg),直至肿瘤发生。治疗使肿瘤发生的中位时间从对照组小鼠的90天延迟至治疗组小鼠的120天。一只接受治疗的小鼠在8月龄后仍未出现肿瘤[2]。 |
| 酶活实验 |
本文介绍了一系列新型非肽类环状膦酸酯和膦酰胺类羟肟酸的设计、合成及其构效关系(SAR)研究,这些化合物是基质金属蛋白酶MMP-1、MMP-3和MMP-9的抑制剂。基于模型研究和X射线衍射分析,我们构建了这些新型化合物在MMP活性位点的结合模型。该模型能够合理解释观察到的SAR数据,包括对不同S1'导向取代基、锌络合基团、手性以及环状膦酸酯和膦酰胺环结构变化的系统研究。我们评估了四种化合物在人纤维肉瘤小鼠模型(HT1080)中的体内活性,并与参考化合物普利诺司他(Prinomastat)进行了比较。所有四种化合物均观察到对肿瘤生长的抑制作用[1]。
明胶酶谱抑制试验:将C57MG/Wnt1细胞的条件培养基进行SDS-PAGE电泳,电泳凝胶中含有1%明胶。电泳后,将凝胶在含有或不含AG3340(20 μg/ml)的缓冲液中于37°C孵育过夜,缓冲液成分为50 mM Tris-HCl(pH 7.5)、5 mM CaCl₂、1 μM ZnCl₂和1% Triton X-100。然后用考马斯亮蓝染色,以显示明胶酶活性,在蓝色背景上呈现清晰的条带。与AG3340孵育的凝胶中未出现清晰条带,证实了MMP蛋白水解活性受到抑制[2]。 |
| 细胞实验 |
Western Blot 分析[1]
细胞类型: C57MG/Wnt1 细胞 测试浓度: 0.1 µg/mL,1 µg/mL 孵育时间: 4 天 实验结果: C57MG/Wnt1 细胞中 MMP-3 启动子活性显著降低。 细胞增殖和细胞迁移实验。[2] 对于 FACS® 分析,将细胞接种于 100 mm 培养皿中,并在有或无 10 µg/ml Prinomastat (AG3340) 的情况下培养至汇合。用 PBS 洗涤细胞,重悬于 70% 乙醇中,并在 4°C 下保存过夜。将细胞与 20 µg/ml RNase A 在 37°C 下孵育 30 分钟,离心后重悬于含 40 µg/ml 碘化丙啶的 PBS 缓冲液中。使用配备 Expo 32(版本 1.2)软件的 EPICS Elite ESP 细胞分选仪进行分析。为评估细胞迁移,用移液器吸头在汇合的 C57MG 或 C57MG/Wnt1 细胞单层上划痕,形成无细胞区域。将细胞在有或无 MMP 抑制剂的条件下孵育 24 小时,并使用配备 Olympus DPII 数码相机的 Olympus IMT-2 显微镜拍摄伤口愈合情况(反映细胞迁移),以此记录伤口愈合过程。对于每种条件,沿伤口选取10个显微视野,并计数从伤口边缘迁移到无细胞区域的细胞。 细胞培养和处理:将小鼠乳腺上皮细胞C57MG及其Wnt1转染衍生物(C57MG/Wnt1)培养于含10%胎牛血清的DMEM培养基中。根据实验[2],用浓度为10 μg/ml或50 μg/ml的AG3340处理细胞,处理时间从24小时到4天不等。 酶谱分析和蛋白质印迹:收集用或不用AG3340(10-50 μg/ml)或rTIMP-2(10 μg/ml)培养的C57MG/Wnt1细胞的条件培养基或细胞裂解液。采用酪蛋白酶谱法分析样品以检测 MMP-3 活性,或采用蛋白质印迹法,使用针对 MMP-3、细胞周期蛋白 D1、磷酸化 Erk1/2 和总 Erk1/2 的特异性抗体进行检测 [2]。 β-catenin 活性报告基因检测 (Topflash):将细胞与 TCF-荧光素酶报告基因 pTopflash 和 Renilla 荧光素酶对照质粒 pRL-SV40 共转染。24 小时后,用 AG3340 (10 μg/ml) 或 NNGH (50 μM) 处理细胞,或不处理,继续培养 24 小时。使用双荧光素酶报告基因检测系统测定荧光素酶活性,结果以萤火虫荧光素酶与海肾荧光素酶活性的比值表示[2]。 MMP-3启动子活性报告基因检测:将细胞与pGL2-SL1-luc(含有驱动萤火虫荧光素酶的鼠源MMP-3启动子)和pRL-SV40共转染。24小时后,用或不用AG3340(50 μg/ml)处理细胞24小时,然后测定双荧光素酶活性[2]。 细胞增殖实验(FACS):将C57MG和C57MG/Wnt1细胞培养至汇合,用或不用AG3340(10 μg/ml)处理24小时。收集细胞,用70%乙醇固定,在RNase A (20 μg/ml)存在下用碘化丙啶 (40 μg/ml)染色,并通过流式细胞术分析细胞周期分布[2]。 细胞迁移实验(伤口愈合):用移液器吸头在汇合的C57MG和C57MG/Wnt1细胞单层上划痕。将细胞在有或无AG3340 (10 μg/ml)或rTIMP-2 (10 μg/ml)的条件下孵育24小时。通过拍照记录细胞向伤口的迁移情况,并在每种条件下计数10个显微镜视野中的迁移细胞数量[2]。 |
| 动物实验 |
本研究旨在评估基质金属蛋白酶合成抑制剂普利诺司他(AG3340)治疗玻璃体内注射分散酶诱导的实验性增生性玻璃体视网膜病变(PVR)的疗效。
将53只新西兰白兔的单眼玻璃体内注射0.07单位分散酶以诱导PVR。PVR诱导一周后,将53只兔子随机分为两组(27:26),分别接受0.5 mg普利诺司他或药物赋形剂(酸化水)的玻璃体内注射,每两周一次。对PVR严重程度进行评分(1-5分),并将普利诺司他治疗组与对照组进行比较。 [4] 肿瘤生物学:人源异种移植研究[3] 在实验动物资源中心开展了人结肠癌、前列腺癌、肺癌、胃癌和鼠黑色素瘤的研究。使用COLO-320DM细胞的研究首先从供体无胸腺小鼠中收集连续传代的肿瘤(约500-1000 mm³),并制备1-2 mm大小的肿瘤组织块,用于移植到未接受过免疫的小鼠体内。肿瘤细胞或组织块双侧植入(每只小鼠2个部位)。使用其他人类细胞系的研究首先从细胞培养物中收集处于指数生长期的细胞,并制备细胞悬液用于皮下移植。肿瘤移植后,小鼠随机分组,耳部打孔以进行识别,并以3只/笼的方式饲养;每项研究包括对照组和普利诺司他(AG3340)治疗组,每组10-12只动物。通常情况下,在开始给予 AG3340 或赋形剂(pH 2.3 的无菌水)给药前,肿瘤需培养 5 天。AG3340 采用无菌 20 g × 1.5 英寸的胃内灌注针进行口服给药。动物每周给药 7 天,每日两次,分别在上午 9 点和下午 4 点左右。因此,对于每日两次给予 100 mg/kg AG3340 的组,每日总剂量为 200 mg/kg。 通过使用电子游标卡尺测量皮下肿瘤的长和宽,并计算肿瘤体积来评估肿瘤生长情况。体积计算采用公式 1/2 (长度)(宽度)²。此外,在整个实验过程中,还评估了给药方案、载体和 AG3340 对小鼠体重和总体健康状况的影响。 研究在卡尔加里大学进行[3] 人 U87 胶质瘤研究已获得机构动物护理和使用委员会的批准。研究开始时,将 5 × 10⁶ 个细胞/位点植入对照组和 Prinomastat (AG3340) 治疗组,每组 5-8 只动物。肿瘤形成 2 至 4 周后,开始腹腔注射载体或 Prinomastat (AG3340)。动物每天给药一次,每周 5 天(MF);周六给予双倍剂量,周日不给药。肿瘤面积根据面积公式(长度)×(宽度)进行测量。 Hoffmann-La Roche公司开展的研究[3] 开展了人MDA-MB-435乳腺癌细胞的研究。研究开始时,将1.5 × 10⁶个细胞植入无胸腺小鼠的乳腺脂肪垫中,分为对照组和Prinomastat (AG3340)治疗组(每组n=10)。待肿瘤形成超过2周后,开始以每周7天的给药方案,每日两次口服给予赋形剂或Prinomastat (AG3340)。肿瘤体积采用公式 1/2 (长度)(宽度) 2 计算。 肿瘤生物学:小鼠静脉注射诱导转移研究 [3] 对数生长期的 B16-F10 细胞用 0.25% 胰蛋白酶-EDTA 从培养物中消化下来,用含胎牛血清 (FCS) 的培养基洗涤,离心,并重悬于无血清培养基中。然后将细胞置于冰上。实验中,将对照组和普利诺司他 (AG3340) 处理组随机分为 12 只/组。使用装有 30G 无菌针头的 1 ml 无菌注射器,将 10⁵ 个 B16-F10 细胞以 0.2 ml 肿瘤体积注入 C57BL/6 小鼠的尾静脉。细胞活力超过 90%,并且在将肿瘤细胞植入小鼠体内所需的 1-2 小时内保持活力。 MMP 抑制对肿瘤形成的影响。[2] 将 MMTV-Wnt1 小鼠进行繁殖,并观察其乳腺肿瘤的形成情况,与每周两次腹腔注射 50 mg/kg 剂量的 Prinomastat (AG3340) 的同窝小鼠进行比较。每周通过触诊监测小鼠乳腺中小肿瘤结节的形成,并记录肿瘤发生的年龄。 MMTV-Wnt1 转基因小鼠的预防性治疗研究:** 使用雌性 MMTV-Wnt1 小鼠(由于 MMTV 启动子驱动的 Wnt1 过表达而发生乳腺肿瘤)。从 6 周龄开始,小鼠每周两次腹腔注射 50 mg/kg 剂量的 AG3340。对照组同窝小鼠未接受任何治疗。每周通过触诊监测小鼠乳腺肿瘤的发生情况。记录肿瘤发生年龄,并使用 Kaplan-Meier 分析比较治疗组和未治疗组的无瘤生存期 [2]。 MMTV-Wnt1 转基因小鼠的预防性治疗研究:本研究使用雌性 MMTV-Wnt1 小鼠(由于 MMTV 启动子驱动的 Wnt1 过表达而发生乳腺肿瘤)。从 6 周龄开始,小鼠每周两次腹腔注射 AG3340,剂量为 50 mg/kg。对照组同窝小鼠未接受任何治疗。每周通过触诊监测小鼠乳腺肿瘤的发生情况。记录肿瘤发生年龄,并使用 Kaplan-Meier 分析比较治疗组和未治疗组的无瘤生存期 [2]。 |
| 药代性质 (ADME/PK) |
生物半衰期
2-5 小时 |
| 毒性/毒理 (Toxicokinetics/TK) |
在体内研究中,接受AG3340(50 mg/kg,腹腔注射,每周两次)治疗的小鼠未表现出任何明显的毒性迹象。该治疗耐受性良好[2]。
|
| 参考文献 |
|
| 其他信息 |
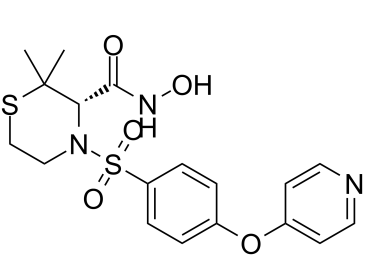
普利诺司他是一种羟肟酸,化学名称为(3S)-N-羟基-2,2-二甲基硫代吗啉-3-甲酰胺,其中硫代吗啉氮原子上的氢原子被[4-(吡啶-4-氧基)苯基]磺酰基取代。它是基质金属蛋白酶(MMPs) 2、3、9、13和14的选择性抑制剂。它具有抗肿瘤活性,可用作MMPs和EC 3.4.24.35(明胶酶B)的抑制剂。普利诺司他属于羟肟酸、硫代吗啉、磺酰胺、芳香醚和吡啶类化合物。它是普利诺司他(1+)的共轭碱。普利诺司他是一种具有潜在抗肿瘤活性的合成羟肟酸衍生物。普利诺司他通过抑制基质金属蛋白酶(MMPs),特别是MMP-2、9、13和14,诱导细胞外基质降解,从而抑制血管生成、肿瘤生长、侵袭和转移。作为一种亲脂性药物,普利诺司他可以穿过血脑屏障。(NCI04)
药物适应症 普利诺司他已被研究用于治疗脑癌、肺癌和前列腺癌。 基质金属蛋白酶(MMPs)在肿瘤进展的后期阶段发挥着明确的作用。然而,有证据表明它们也参与了恶性转化的早期阶段。Wnt信号通路在许多上皮癌的发生发展中起着至关重要的作用。本研究利用Wnt1诱导的C57MG小鼠乳腺上皮细胞上皮-间质转化(EMT)来探讨MMPs在恶性肿瘤早期进展中的作用。在C57MG细胞中过表达Wnt1可促进EMT、β-catenin从细胞膜向细胞核的转位及其转录活性、细胞增殖和细胞迁移。同时,我们观察到,与不表达Wnt1的C57MG细胞相比,表达Wnt1的C57MG细胞中基质金属蛋白酶-3(MMP-3)的表达增加,且MMP-3启动子活性提高了5.5倍。用MMP抑制剂AG3340处理Wnt1过表达细胞后,MMP-3的表达降低。我们还发现,MMP-3和Wnt3a协同增强了C57MG细胞中β-catenin的转录活性。与此一致,MMP抑制剂或siRNA下调MMP-3后,Wnt1对EMT、增殖和迁移的影响受到抑制。这些结果表明,MMP-3既是Wnt/β-catenin信号通路的直接转录靶点,也是该通路的重要组成部分。[2] Prinomastat (AG3340)是由Agouron/辉瑞公司开发的一种合成的广谱基质金属蛋白酶抑制剂。本研究将其作为药理学工具,探讨MMP在Wnt1诱导的上皮-间质转化和乳腺肿瘤发生中的作用。结果表明,AG3340抑制MMP可以逆转EMT,减少细胞增殖和迁移,并在体外下调β-catenin的转录活性。此外,在MMTV-Wnt1转基因小鼠中预防性给予AG3340可延缓乳腺肿瘤的发生,这支持了MMPs积极参与Wnt介导的转化以及MMP抑制可能是一种癌症预防策略的观点。这些发现揭示了MMPs在肿瘤进展早期阶段的复杂作用,超越了其在侵袭和转移中众所周知的功能[2]。 |
| 精确质量 |
423.092
|
|---|---|
| 元素分析 |
C, 51.05; H, 5.00; N, 9.92; O, 18.89; S, 15.14
|
| CAS号 |
192329-42-3
|
| 相关CAS号 |
Prinomastat hydrochloride;1435779-45-5
|
| PubChem CID |
466151
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.377g/cm3
|
| 折射率 |
1.615
|
| LogP |
4.123
|
| tPSA |
146
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
28
|
| 分子复杂度/Complexity |
638
|
| 定义原子立体中心数目 |
1
|
| SMILES |
O=C([C@@H]1N(S(=O)(C2=CC=C(OC3=CC=NC=C3)C=C2)=O)CCSC1(C)C)NO
|
| InChi Key |
YKPYIPVDTNNYCN-INIZCTEOSA-N
|
| InChi Code |
InChI=1S/C18H21N3O5S2/c1-18(2)16(17(22)20-23)21(11-12-27-18)28(24,25)15-5-3-13(4-6-15)26-14-7-9-19-10-8-14/h3-10,16,23H,11-12H2,1-2H3,(H,20,22)/t16-/m0/s1
|
| 化学名 |
(S)-N-hydroxy-2,2-dimethyl-4-((4-(pyridin-4-yloxy)phenyl)sulfonyl)thiomorpholine-3-carboxamide
|
| 别名 |
AG3340; AG-3340; AG 3340; KBR-9896; CHEMBL75094; (3S)-N-hydroxy-2,2-dimethyl-4-(4-pyridin-4-yloxyphenyl)sulfonylthiomorpholine-3-carboxamide; 10T6626FRK;KBR 9896; KBR9896; Prinomastat.
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO :< 1 mg/mL
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00004200 | COMPLETED | Drug: prinomastat Drug: temozolomide |
Brain and Central Nervous System Tumors | Pfizer | 1999-10 | Phase 2 |
| NCT00004199 | COMPLETED | Drug: cisplatin Drug: gemcitabine hydrochloride Drug: prinomastat |
Lung Cancer | Pfizer | 1999-03 | Phase 3 |
| NCT00003343 | COMPLETED | Drug: endocrine-modulating drug therapy Drug: mitoxantrone hydrochloride Drug: prednisone Drug: prinomastat |
Prostate Cancer | Pfizer | 1998-03 | Phase 3 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
