| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
IC50: 0.069 μM (GluN2C), 0.035 μM (GluN2D), 5.2 μM (GluN2A), 16 μM (GluN2B)[1]
|
|---|---|
| 体外研究 (In Vitro) |
研究人员用甘氨酸和抑制剂2i (DQP-997-74)对细胞进行预孵育,并比较了谷氨酸应用时抑制剂在峰值和稳态时的效力。抑制剂的预孵育产生了峰值电流的降低,这表明2i可以在没有谷氨酸的情况下与受体结合,在谷氨酸结合和通道被激活的那一刻抑制峰值电流。化合物2i在峰值电流响应时的IC50为0.23 μM,是稳态响应时的3倍(IC50为0.08;图4 b, C;表2),表明谷氨酸结合后对2i的亲和力增加,这使得2i与NMDAR重新平衡,产生时间依赖性抑制,反映了抑制剂结合的时间过程。为了研究2i抑制的时间过程,我们用单指数函数拟合了当前反应的弛豫。如果电流响应时间过程反映2i结合,则τ抑制的倒数应与其浓度线性相关,斜率等于kON,截距等于kOFF。电流响应的指数时间过程与化合物2i浓度相关(R2 = 0.82),线性回归分析得出kON的KD值为4.0 × 105 M-1 s-1, kOFF的KD值为0.17 s-1(图4D),表明KD值为0.43 μM。当浓度为0.1 μM时,化合物2i的存在减缓了GluN1/GluN2D受体的失活(5.3±0.2 s);p = 0.017)和0.3 μM(10.5±1.0 s);p = 0.002),但在0.03 μM时不存在(5.6±0.3 s);P = 0.518;图4 e)。这些数据表明,在2i的存在下,谷氨酸的解结合速度减慢。这种现象之前在QNZ-46中被发现,QNZ-46是一种谷氨酸依赖GluN2C/ 2d选择性抑制剂,具有不同的结构核心正如该研究提出的那样,长时间失活可能表明2i减缓了谷氨酸解结合所必需的构象变化,或者在谷氨酸解结合之前,2i可能需要解结合。[1]
研究人员还通过比较谷氨酸应用的峰值和稳态时抑制剂的效力,评估了GluN1/GluN2C在HEK细胞中表达的抑制机制,以抑制抑制剂2i (DQP-997-74)。在峰值电流响应时,化合物2i在GluN1/GluN2C受体上的IC50 (IC50 5.0 μM, 95%置信区间为1.4 ~ 8.5 μM)高于稳态响应时的IC50 (IC50 0.25 μM, 95%置信区间为0.15 ~ 0.35 μM;F(1,50) = 81.34, p < 0.0001;见图S2),表明与没有谷氨酸结合的NMDARs相比,2i在谷氨酸结合的GluN1/GluN2C受体上的亲和力增加。电流响应的指数时间过程与化合物2i浓度相关(R2 = 0.79),线性回归分析得出kON的KD值为8.0 × 105 M-1 s-1, kOFF的KD值为0.27 s-1(图4D),表明KD值为0.33 μM。当浓度为1 μM时,化合物2i的存在使GluN1/GluN2C受体失活(0.53±0.07 s)减慢(1.3±0.14 s);p = 0.001)和3 μM(2.3±0.40 s);p < 0.0001),但在0.1 μM时不存在(0.61±0.07 s);p = 0.965)和0.3 μM(0.88±0.13 s;P = 0.115;图S2)。 [1] 此外,研究人员评估了谷氨酸依赖如何影响对短暂激动剂应用的抑制反应,类似于发生在突触的阶段性谷氨酸释放。为此,我们用抑制剂2i对表达GluN1/GluN2D受体的细胞进行预孵育,然后在加压微移液管中应用5个短脉冲的NMDA(比谷氨酸解离更快)和甘氨酸来模拟重复突触传递(图4F)。我们测量了峰幅,并比较了第一反应和最后反应的抑制浓度-反应曲线(图4G)。在没有药物控制的情况下,对诸如受体内化等系统性错误的初始脉冲序列。在短暂的激动剂应用过程中,化合物2i在第一次脉冲(IC50 = 0.75 μM)时的IC50比第5次脉冲(IC50 = 0.40 μM;图4F,G),这表明在长时间或重复的受体激活过程中,抑制可能更大。 [1] 为了进一步评估2i是否遵循质量作用规律,研究人员在HEK细胞中表达的GluN1/GluN2D受体的稳态谷氨酸和甘氨酸反应中,将不同浓度的2i与谷氨酸共应用(图4H)。我们测量了所测试的每种浓度的2i产生的稳态抑制作用,并确定在这些条件下化合物2i的IC50为0.10 μM(图4I, Hill slope 0.93),与预施用时稳态的IC50相似(图4B)。我们还用单一指数函数拟合了化合物2i产生的抑制的开始和抵消时间过程(图4J)。电流响应的指数时间过程与化合物2i浓度相关(R2 = 0.51),线性回归分析得出kON和kOFF的KD值分别为2.1 × 105 M-1 s-1和0.05 s-1, KD值为0.24 μM。正如预期的那样,从2i抑制中恢复的时间过程与药物与单一位点结合的浓度无关,这表明2i的抑制并不涉及增强脱敏。 |
| 体内研究 (In Vivo) |
DQP 2iDQP-997-74>对Tsc1±小鼠癫痫发作[1]
的影响 研究了2i/DQP-997-74DQP-997-74DQP-997-74DQP-997-74 dqp -997对雄性Tsc1±小鼠体内癫痫发作的急性作用。Tsc1±小鼠GluN2C.41表达上调14 mg/kg剂量的2i显著减少了新皮层2/3层和4层的自发电图癫痫发作(图6A)。在一组4只小鼠中,注射2i剂量后,两只小鼠的癫痫发作完全停止,而其余小鼠在注射后约70分钟后无癫痫发作。此外,当剂量增加到28 mg/kg时,癫痫事件立即停止,与对照组相比,癫痫发作频率、持续时间和幅度均显著下降(图6B,C)。在一组5只Tsc1±小鼠中,28 mg/kg剂量的2i完全停止了4只受试者的癫痫发作事件,而最后一只小鼠在约50分钟后表现出可观察到的癫痫发作减少。这种体内抗癫痫活性说明了该类化合物对GluN2C/ d相关癫痫的治疗潜力。 <人力资源> 前药的体内评价[1] 接下来,我们通过比较静脉给药前药或母体化合物2i后的脑和血浆2i浓度,研究了前药策略是否可以改善化合物2i/DQP-997-74的脑/血浆比率。尽管酯类前药在体外稳定性较低,但我们测试了前药211、2m、2o和2p的药代动力学分析是否可以作为未来前药开发的概念证明。母体化合物2i静脉给药15min时血药浓度和脑药浓度峰值分别为642和14 ng/mL,随后迅速降低。血浆中2i的水平在1小时内下降了约10倍,脑组织中2i的水平在3小时内下降了BLQ(图5B)。几种前药增加了母体化合物2i的浓度和在脑内停留的时间(图7)。前药在血浆和脑内的峰值水平分别为1190/119 ng/mL(化合物2l)、786/64 ng/mL(化合物2m)、563/26 ng/mL(化合物2o)和647/56 ng/mL(化合物2p)。所有前药静脉给药后脑内2i水平均高于单独给药后脑内2i水平。给药后1小时,前药组2i脑内水平比单独给药组2i高5 - 10倍。在3h时,未检测到2i给药的脑水平,而三种前药在3h时具有合理的脑/血浆比,Kp值分别为1.36 (2l), 0.36 (2m)和0.64 (2p),表明前药比2i暴露更高,持续时间更长。与母体2i相比,2i脑/血浆比值的增加是由于在静脉给药后,2i在大脑中的含量增加,在大脑中停留的时间更长(图7)。尽管在向2a中添加两种氟和一种酯后,脑暴露仍然很低,这些前药的微摩尔浓度对CYP3A4产生了一定的抑制(表5)或进行了快速代谢,然而,这些数据表明,前药方法可以提高大脑的浓度。如果能够设计出在血浆和肝微粒体中更稳定的前药基团,并且与本文探索的简单酯相比,具有更好的血脑屏障穿透性,那么前药策略可能会提供更强大的工具化合物和潜在的临床先导系列。 |
| 酶活实验 |
肝微粒体稳定性[1]
人肝微粒体(HLMs, 20 mg/mL)和CD-1小鼠肝微粒体(MLMs, 20 mg/mL)在10 mM的蒸馏水 原液中制备。维拉帕米和苯海拉明分别作为HLM和MLM稳定性的阳性对照。试验化合物和阳性对照最初溶解在DMSO中,制成10 mM的原液。然后将样品溶液用70% MeOH/H2O或100% men进一步稀释至500 μM。然后,将人或小鼠肝微粒体(55 μL)与磷酸钾缓冲液(100 mM, 928 μL)在1.5 mL埃彭多夫管中混合制备反应。将实验化合物(6.6 μL 500 μM溶液)加入到悬浮液中,在37℃下孵育5 min,然后用110 μL 10 mM NADPH引发肝微粒体反应,在37℃下孵育指定时间。实验条件为:终体积为1100 μL(有机溶剂含量<0.6%),高通量质粒和传销质粒浓度为1 mg/mL,终测试化合物浓度为3 μM。每隔0、5、10、15、30 min,从每个反应混合物中取出等量(100 μL),用100 μL冷内标溶液(ISTD, 2 μM 7-乙氧基- 5-香豆素在MeOH或MeCN中)淬灭。然后将冷却后的等分液在12 500 g下离心5-10 min,取出所得上清液,置于LC-MS小瓶中,用LC-MS/MS (Agilent G6460C QQQ质谱联用Infinity II 1260高效液相色谱)进行分析。为保证质量,对每种化合物进行了正、负对照试验。阳性对照反应在每个时间点的终体积为550 μL时进行。最后,在不含NADPH (150 μL)的情况下,用被试化合物和肝微粒体进行阴性对照实验,并在30 min时间点进行分析。 等离子体稳定性[1] 人血浆(锂肝素(LiHep)混合,性别混合,0.2 μm过滤)和小鼠血浆(BALB/C, LiHep混合,男性混合,0.2 μm过滤)购自BioIVT。普鲁卡因在人和小鼠血浆实验中均为阳性对照。试验化合物和阳性对照最初溶解在DMSO中,制成10 mM的原液。然后用70%的MeOH/H2O或100%的men将被测化合物和对照化合物的溶液进一步稀释至500 μM。接下来,将人或小鼠血浆(994 μL)引到1.5 mL埃彭多夫管中,每种化合物制备重复反应(反应A和反应B)。等离子体在37°C下孵育10 min,然后加入6 μL 500 μM溶液的测试化合物引发反应,并在37°C下继续孵育指定的时间。该方法提供了最终体积为1000 μL(有机溶剂含量<0.6%),最终测试化合物浓度为3 μM的重复实验。分别在0、15、30、60和120 min的时间间隔内从每个反应混合物中取出等量(100 μL),用150 μL的冷ISTD溶液(2 μM 7-乙氧基- 5-香豆素在MeOH或MeCN中)淬灭。然后将淬灭的等分液在15,000 g离心30-45分钟,提取所得上清液(~ 70 μL),置于LC-MS小瓶中,用LC-MS/MS (Agilent G6460C QQQ MS与Infinity II 1260高效液相色谱联用)进行分析。为保证质量,对每种化合物进行了正、负对照试验。阳性对照反应在每个时间点的终体积为1000 μL,单次运行。最后,用143 μL的DPBS中被试化合物进行阴性对照实验,并在120 min时间点进行分析。 脱靶动作分析[1] 在0.5M CGP-78608的存在下,化合物(S)-(-)-2i在1M检测对甘氨酸激活的GluN1/GluN3A(人GluN1- f484a,T518L/GluN3A指GluN1- fa,TL/GluN3A)和大鼠GluN1-4a/GluN3B的作用。GluN1/GluN3的cRNA注射比例为1:2。还检测了化合物(S)-(-)- 2i对爪蟾卵母细胞中表达的AMPA(大鼠GluA1、大鼠GluA2、人GluA3-L531Y)、kainate(大鼠GluK2)、烟碱乙酰胆碱(大鼠7和42,cRNA比例1:1)、GABAA(大鼠S, cRNA比例1:1)、GABAC(大鼠1)、甘氨酸(大鼠1)和嘌呤能(人P2X)受体的作用。利用mMessage mMachine试剂盒从线性模板cDNA合成编码受体亚基的cRNA。卵母细胞注射1-5 ng cRNA,体积为50 nL,在15°C的Barth溶液中孵育2-5天,然后进行记录。表达gluk2的卵母细胞在1 mg/ml(10M)的刀豆蛋白a中孵育10分钟后进行记录。编码GABAC和甘氨酸亚基的cdna由Dr. D. Weiss提供。编码烟碱乙酰胆碱受体亚基的cdna由dr。R. Papke和S. Heinemann。编码嘌呤能受体的cdna由Dr. R. Hume提供。100M GABA激活GABAA和GABAC受体。以指定浓度的乙酰胆碱(μM)激活烟碱乙酰胆碱受体α7(300)。100M甘氨酸用于激活甘氨酸受体,100M甘氨酸用于激活GluN1/GluN3受体,100M谷氨酸用于激活AMPA和kainate受体,9M ATP用于激活嘌呤能受体。测试化合物存在时的反应以对照的百分比表示。 代谢稳定性测定方法[1] 采用Agilent 1260 Infinity II高效液相色谱(HPLC)和Agilent G6460三重四极杆质谱仪进行LC-MS/MS分析。采用Agilent InfinityLab Poroshell 120 EC-C18 (2.1 x 50 mm, 2.7m)或EC-C8 (2.1 x 50 mm, 2.7m)色谱柱在40℃下进行反相HPLC S10分离。流动相为甲醇-水(0.1% FA)或甲醇-水(0.1% FA),流速为0.5 mL/min。每种方法均在内标(ISTD) d5-7-乙氧基香豆素存在下建立。采用多反应监测(MRM)模式,采用安捷伦喷射流电喷雾正离子(ESI+)进行前驱体和产物离子检测。单个化合物的所有MRM跃迁、碎片电压和碰撞能量在补充表S4中提供。其他标准质谱条件为:静置时间100ms;气体流量10l /min;雾化器压力45psi;EMV为200v。最后,使用Agilent 6460定量分析软件对所有采集的数据进行处理。 动力学溶解度实验方法[1] 首先将测试化合物溶解在DMSO中,形成30 mM或60 mM的原液。然后在96孔微滴板中进行动力学溶解度实验,每孔最终体积为250µL,排在B-H。每个板初始制备时,在A3孔中添加16 μL DMSO,在A4孔中添加20 μL,在A5孔中添加27 μL,在A排(A1和A6-12)其余孔中添加40 μL。将实验化合物(DMSO原液30 μL)直接加入A2孔,不作进一步修饰。将实验化合物(DMSO原液64 μL)在A3孔中稀释,使其总体积为80 μL。然后进一步稀释,从A3井中取出60µL加入到A4中,然后从A4井中取出53µL加入到A5中,然后从A5井中取出40µL加入到A6中,依次从其余井中取出40µL,直到A12。A1作为DMSO空白。将A行制备的样品溶液(每孔2.5µL)转移到B-H行相应的孔中。杜尔贝科的磷酸盐(DPBS 1 x,没有Ca2 +和Mg2 +, S11 pH值7.1 - -7.3)被添加到所有井行b - h(30µL)和96孔板在25摄氏度孵化1分钟。接下来,DPBS(217.5µL)被添加到所有井行b - h和96孔板在25摄氏度再次孵化2 h。这个过程为七个复制实验提供了最终的测试化合物的浓度(1% DMSO DPBS)从300到600年0.9375µM或b - h 1.875µM行,取决于是否使用30mm或60mm DMSO原液(即从a行样品制备中稀释100倍)。孵卵后,使用NEPHELOstar®微孔板读取器分析,并使用MARS分析软件处理分段线性回归数据。 |
| 细胞实验 |
NMDA受体的电压箝位分析[1]
双电极电压钳记录[1] 未受精的非洲爪蟾卵母细胞从Ecocyte公司获得,或从爪蟾1号(Dexter, MI)购买卵巢,卵母细胞如前所述制备。42,47对表达重组大鼠GluN1/GluN2A、GluN1/GluN2B、GluN1/GluN2C或GluN1/GluN2D的卵母细胞进行双电极电压钳记录。大鼠GluN1-1a(简称GluN1;NCBI参考序列NM_017010.2)、GluN2A (NM_012573.4)、GluN2B (NM_012574.1)、GluN2C (NM_012575.3)和GluN2D (NM_022797.2)由Salk研究所的S. Heinemann博士、京都大学的S. Nakanishi博士和海德堡大学的P. Seeburg博士提供。卵母细胞分离、cRNA合成和cRNA注射如前所述进行。32,33,47简单地说,将5-10 ng的cRNA注入到不含rnase的水中,GluN1:GluN2的比例为1:1 ~ 1:5。卵母细胞在15-19℃的Barth 's溶液中孵育,该溶液由(mM) 88 NaCl、1 KCl、2.4 NaHCO3、10 HEPES、0.82 MgSO4、0.33 Ca(NO3)2和0.41 CaCl2组成,并添加100 μg/mL庆大霉素、40 μg/mL链霉素和50 μg/mL青霉素。注射细胞外记录液后2-4天进行记录,细胞外记录液含有(mM) 90 NaCl、1 KCl、10 HEPES、0.5 BaCl2和0.01 EDTA, pH为7.4,用NaOH调节。为了防止电流反应在实验过程中逐渐增加,一些表达GluN1/GluN2A的卵母细胞被注射20-50 nL的2-50 mM K-BAPTA。以谷氨酸(100 μM)和甘氨酸(30 μM)为最大有效浓度,以谷氨酸和甘氨酸为最大有效浓度(30 μM),以谷氨酸和甘氨酸为最大有效浓度(30 μM),得到测试化合物的浓度-响应曲线。在DMSO中配制20 mM的原液,在记录液中稀释至终浓度。DMSO含量为0.05 ~ 0.5% (v/v)。在2、5或10 mM羟丙基- β -环糊精中测试了一些IC50值为μM的低效化合物(2l、2o、2p和2r)。在辅助资料中给出了脱靶分析的方法。 全细胞电压钳记录[1] 将HEK293细胞(HEK, ATCC CRL-1573)镀于0.1 mg/mL聚赖氨酸预处理的玻璃罩上,在添加10%胎牛血清、10 U/mL青霉素和10 μg/mL链霉素的Dulbecco 's modified Eagle培养基(DMEM)中培养,37℃,5% CO2。48 .采用磷酸钙沉淀法,以1:1:5的比例瞬时转染编码大鼠GluN1、GluN2D和eGFP的cDNA转染24-48 h后,用外用记录液灌注细胞,记录液中含有(mM) 3 KCl、150 NaCl、0.01 EDTA、1.0 CaCl2、10 HEPES和22 d-甘露醇(NaOH调节pH至7.4)。贴片电极(电阻3-5 MΩ)由薄壁玻璃微移液器通过双级玻璃微移液器(PC-10)制备,内填充含有(mM) 110 d-葡萄糖酸盐、110 CsOH、30 CsCl、5 HEPES、4 NaCl、0.5 CaCl2、2 MgCl2、5 BAPTA、2 NaATP和0.3 NaGTP的溶液。用CsOH调节pH至7.4;渗透压调整到300 - 305mosmol /kg左右。使用最大有效浓度的激动剂(100 μM谷氨酸和30 μM甘氨酸)在−60 mV保持电位下激发全细胞电流响应,并使用Axopatch 200B膜片钳放大器记录。双管玻璃微移液管由压电转换器控制,用于溶液的快速交换。在一些实验中,500 μM NMDA和250 μM甘氨酸通过膜片移液器短暂加压(提示3-5 MΩ)。电流响应在2 kHz低通滤波,8极贝塞尔滤波器(- 3 dB),并使用由Clampex 10.3控制的Digidata 1440A采集系统(Molecular Devices)在20 kHz数字化。所有贴片实验均在室温(23℃)下进行。 |
| 动物实验 |
IP Administration [1]
A group (n = 15) of fed, male C57BL/6 mice ∼6 to 8 weeks of age was injected IP with 10 mg/kg (1 mg/mL) of drug using 50% PEG400 in water as a vehicle. Samples were collected from the blood and brain at 0.25, 0.5, 1, 2, and 4 h after administration (3 mice per time point) following CO2 anesthesia. Collection from the brain was performed as follows: the mouse is terminally anaesthetized via rising concentration of CO2 and as much blood is removed as possible via cardiac puncture. The cardiac puncture is done by opening the chest cavity to expose the heart, cutting an incision in the right auricle using surgical scissors, and finally injecting a saline solution (∼10 mL) slowly into the left ventricle via a syringe. The mouse is placed head down at a 45° angle to facilitate blood removal. After perfusion, the skull is opened and the brain is removed. The whole brain is washed with saline, dried with surgical gauze, placed in tared tubes, and stored at −75 °C before analysis. For plasma sample preparation at the 0.25–2 h time points, 15 μL of blank solution, 30 μL of plasma sample, and 150 μL were added sequentially for protein precipitation. After centrifugation, 20 μL of the supernatant and 80 μL of collection buffer were combined, and thus, the final compound concentration in plasma (ng/mL) was corrected by multiplying by 5. For plasma sample preparation at the 4 h time point, 15 μL of blank solution, 30 μL of plasma sample, and 150 μL of acetonitrile were added sequentially for protein precipitation. For all brain sample preparation, 15 μL of blank solution, 30 μL of brain samples, and 150 μL of acetonitrile were added sequentially for protein precipitation. Brain samples were prepared by adding brain (g) to deionized water (mL) in a 1:4 ratio for homogenization. The mixtures were then vortexed for 30 s and subsequently centrifuged (∼4000 rpm) for 15 min. The supernatant was diluted 3-fold with water, and a 2 μL aliquot of the diluted supernatant was injected into a Shimadzu LC-30A LC-MS/MS system with a Phenomenex 2.6 μ PFP 100A column (30 mm × 2.1 mm) using verapamil as an internal standard. A gradient from 95% water (0.1% formic acid) to 95% ACN (0.1% formic acid) was run over 2 min at a flow rate of 0.6 mL/min. Brain and blood samples were collected at 15, 30, 60, 120, and 240 min from three C57Bl/6 mice at each time point. See Tables S6–S10 for 2i/DQP-997-74 concentrations. IV Administration [1] Nine male C57BL/6 mice were administered intravenously (IV) with solution formulation of prodrug at 5 mg/kg dose (5 mL/kg) in 5% N-methyl-2-pyrrolidine, 5% Solutol HS-15, and 90% saline. Blood samples (∼60 μL) were collected under light isoflurane anesthesia from a set of three mice at 0.25, 1, and 3 h. Plasma was harvested by centrifugation of blood and stored at −70 ± 10 °C until analysis. Immediately after the collection of blood, brain samples were collected from each mouse at respective time points. Brain samples were homogenized using ice-cold phosphate-buffered saline (pH 7.4), and homogenates were stored below −70 ± 10 °C until analysis. The total homogenate volume was 3 times that of the brain weight. The plasma and brain concentration–time data of compounds 2i/DQP-997-74 were used for the pharmacokinetic analysis. Plasma and brain samples were quantified by the fit-for-purpose LC-MS/MS method (LLOQ: 2.06 ng/mL for plasma and 5.16 ng/mL for brain). Tables S11 and S12 show the 2i/DQP-997-74 concentrations. Acute Epileptic Mouse Model [1] All experiments were conducted in agreement with the requirements of the European Directive 2010/63/EU. Mice were housed in ventilated, light-tight, sound-isolated chambers under a standard 12:12 light/dark cycle (light on at 07.00 PM and light off at 07.00 AM) with food and water available ad libitum. C57BL/6J wild-type (Tsc1+/+) females from Janvier Laboratories (France) were crossed with heterozygote Tsc1± male mice with the genetic background B6;129S4-Tsc1tm1.1Djk/Nci. The genotyping of pups issuing from this cross-breeding was performed on tail tissue samples at postnatal day P9. The study was conducted in Tsc1± male mice at P14–16. Pups from at least three deliveries for each condition were studied to minimize the potential sampling bias. The DQP derivative 2i/DQP-997-74 was diluted in DMSO at a concentration of 100 mM before being added to 100 to 150 μL of saline solution and intraperitoneally injected in mice. Intraperitoneal injection was administered during the EEG recordings at three different concentrations: 7, 14, and 28 mg/kg to estimate the dose–response relationship. Experiments were performed on postnatal days of P14 and P16 male Tsc1± mice. Surgery was performed under isoflurane anesthesia and lidocaine analgesia. During recordings, the head was fixed to the frame of a stereotaxic apparatus by attached bars; animals were surrounded by a cotton nest and heated via a thermal pad (36.6–37.7 °C). A silver chloride reference electrode was placed in the cerebellum. EEG recordings were performed with nonanaesthetized head-restrained Tsc1± mice. A 16 site linear silicon probe (100 μm separation distance between recording sites) was placed into the somatosensory cortex using the Paxinos and Franklin atlas (2001)54 at coordinates anterior–posterior = 2–2.5 mm, mediolateral = 2–3 mm from Bregma, 1.2–1.5 mm from the dural surface, in order to trace the columnar activity at all layers. Signals were amplified (×100) and filtered at 3 kHz using a 16-channel amplifier, digitized at 10 kHz, and then saved to the hard disk of a PC using Axoscope software (Molecular Devices). Recordings were analyzed offline using Clampfit and Origin software. After recording, the position of the silicone probe was verified visually by DiI staining of the electrode in 100 μm coronal sections from the fixed brain. We considered that multiunit activity occurred in epileptic discharges if they appeared in a group of multiple spikes whose amplitude exceeded the background activity within a period lasting for at least 20 s. During EEG recordings, animals were monitored visually to determine the behavioral correlates of each electrographic epileptic discharge. During the experiment, EEG recordings were performed for 2 h prior to drug administration, after which drug was intraperitoneally injected with dosages of 7, 14, or 28 mg/kg to investigate its acute effects on in vivo epileptic seizures. EEG recordings then were performed for 2–2.5 h post injection. |
| 药代性质 (ADME/PK) |
In Vivo Pharmacokinetic Evaluation of 2i/DQP-997-74 and 2k [1]
In order to determine whether 2i (DQP-997-74) and the uncharged amide analogue 2k could be used to probe GluN2C and GluN2D actions in vivo, we performed a mouse pharmacokinetic study and measured the exposure of 2i in plasma and brain tissue (Figure 5). The concentration of 2i observed in the brain remained low with a Cmax of only 23 ng/g after a 10 mg/kg intraperitoneal (IP) injection. When injected intravenously (IV) 2i exhibited even lower Cmax within the brain (14 ng/g at 15 min) and plasma (642 ng/mL at 15 min) that rapidly diminished over time. However, 2i plasma and brain concentrations of 2i remained relatively stable for at least 2 h following IP administration. Compound 2k replaces the carboxylate with an amide, which will be uncharged at physiological pH. Compound 2k showed a similar profile, with modestly lower plasma and brain levels (Table 3), which indicated that removal of the charged carboxylate did not improve the brain permeability. Although mouse brain Cmax levels for 2i were low following IP administration, they were maintained over the course of 4 h, and the compound could reach brain concentrations (40 nM in brain compartment, upper limit of 200 nM if 2i is restricted to extracellular space) that might provide some NMDAR antagonism (GluN2D IC50 130 nM), although the free fraction of 2i in brain (<0.1%) was not improved compared to 2a (Table S2). In Vitro DMPK Profiles for DQP Prodrugs [1] Anticipating that the carboxylate side chain may hinder BBB penetrance, we synthesized and biologically screened a series of 2i/DQP-997-74 prodrugs with ester and amide masking groups. With the classical physiochemical parameters required for brain exposure (i.e., molecular weight, MW, lipophilicity, and topological polar surface area, TPSA) and drug release mechanisms beyond the BBB in mind, we focused on simple alkyl-based promoieties. Our selected lead compound (2i), a close analogue (2a), and their corresponding prodrugs (2l–r) were studied in vitro to ascertain drug solubility, metabolic stability in liver microsomes (LMs), plasma stability, mouse brain homogenate stability, and cytochrome P450 (CYP450) inhibition (Tables S3 and S4 for predicted absorption, distribution, metabolism, and excretion or ADME parameters). We first determined the solubility of compounds 2a, 2i, and 2k–2r in phosphate-buffered saline (PBS, 1% DMSO) at room temperature (Table 4 and Figure S4). Our previous lead 2a demonstrated a relatively high PBS solubility of ∼208 μM. However, we observed a reduction in the solubility of difluoro derivatives 2i (∼38 μM) and 2k (∼30 μM). An accurate solubility comparison via a nephelometer between the active parent species (2i and 2k) and their analogous prodrugs (2l–2r) could not be obtained. However, since all compounds precipitated within the range 25–55 μM, they likely possess aqueous solubility values similar to those of the parent. Compounds 2a and 2i/DQP-997-74 demonstrated high stability (i.e., greater than 95% remaining over the assay time course) in human and mouse LMs and plasma (Table 4 and Figures S5 and S6). Additionally, 2a and 2i either weakly inhibited or displayed no activity against the major drug metabolizing enzymes of CNS clinical relevance, CYP2D6 and CYP3A4 (Table 5 and Figure S4). While amide 2k was stable in both LMs and human plasma, the compound was metabolized in mouse plasma (42% of prodrug after 2 h) and brain homogenate (56% of prodrug after 4 h; Table 4) and moderately inhibited CYP3A4 (IC50 = 2 μM) at concentrations 10-fold higher than the IC50 at GluN1/GluN2D (Table 5). Further metabolite identification experiments are required to elucidate which mouse enzymes degraded 2k. Despite 2k being stable to human and mouse LMs (greater than 90% of parent remaining after 30 min), 2q and 2r decomposed quite rapidly (t1/2 ≤ 7 min; Table 4). Although there is evidence for 2q instability in brain homogenate (33% prodrug remaining after 4 h), 2i, the amidase-catalyzed hydrolysis product, was unobservable and below the limit of quantification (BLQ) by LC-MS/MS (Table 4). Interestingly, 2r was stable under the same conditions (93% prodrug remaining). Since neither the amide moiety produced 2i in brain homogenate nor was stable to hepatic metabolism (presumably from N-demethylation), we did not advance 2q or 2r to in vivo pharmacokinetic studies. [1] All ester prodrugs hydrolyzed to 2i/DQP-997-74 in mouse brain homogenate, with the less sterically hindered promoieties 2l, 2n, and 2p cleaving more efficiently than the bulkier substrates 2m and 2o (Tables 4 and S5). Although stable in human plasma, the ester series also exhibited high metabolic instability in LMs (<5 min) and mouse plasma (<40 min). Additionally, while the ester prodrugs did not inhibit CYP2D6, compounds 2l–o displayed increased CYP3A4 inhibition (6–12 μM) relative to 2i, which suggests the potential for drug–drug interactions (Table 5). |
| 参考文献 |
[1]. D'Erasmo MP, et al. Development of a Dihydroquinoline-Pyrazoline GluN2C/2D-Selective Negative Allosteric Modulator of the N-Methyl-d-aspartate Receptor. ACS Chem Neurosci. 2023 Sep 6;14(17):3059-3076..
|
| 其他信息 |
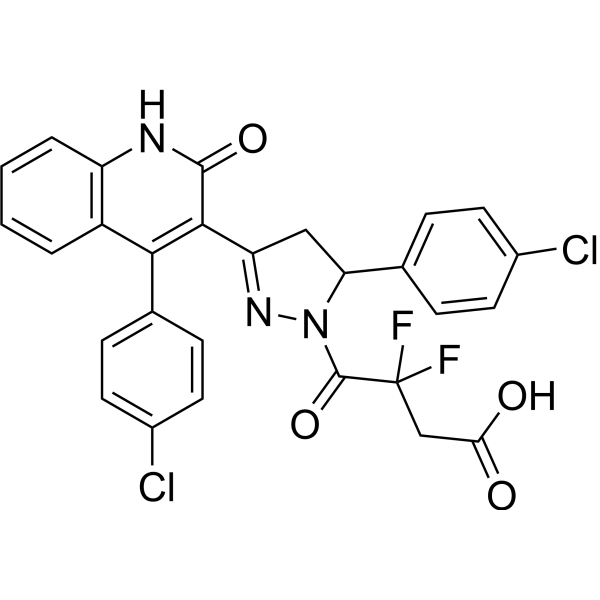
Subunit-selective inhibition of N-methyl-d-aspartate receptors (NMDARs) is a promising therapeutic strategy for several neurological disorders, including epilepsy, Alzheimer's and Parkinson's disease, depression, and acute brain injury. We previously described the dihydroquinoline-pyrazoline (DQP) analogue 2a (DQP-26) as a potent NMDAR negative allosteric modulator with selectivity for GluN2C/D over GluN2A/B. However, moderate (<100-fold) subunit selectivity, inadequate cell-membrane permeability, and poor brain penetration complicated the use of 2a as an in vivo probe. In an effort to improve selectivity and the pharmacokinetic profile of the series, we performed additional structure-activity relationship studies of the succinate side chain and investigated the use of prodrugs to mask the pendant carboxylic acid. These efforts led to discovery of the analogue (S)-(-)-2i, also referred to as (S)-(-)-DQP-997-74, which exhibits >100- and >300-fold selectivity for GluN2C- and GluN2D-containing NMDARs (IC50 0.069 and 0.035 μM, respectively) compared to GluN2A- and GluN2B-containing receptors (IC50 5.2 and 16 μM, respectively) and has no effects on AMPA, kainate, or GluN1/GluN3 receptors. Compound (S)-(-)-2i is 5-fold more potent than (S)-2a. In addition, compound 2i shows a time-dependent enhancement of inhibitory actions at GluN2C- and GluN2D-containing NMDARs in the presence of the agonist glutamate, which could attenuate hypersynchronous activity driven by high-frequency excitatory synaptic transmission. Consistent with this finding, compound 2i significantly reduced the number of epileptic events in a murine model of tuberous sclerosis complex (TSC)-induced epilepsy that is associated with upregulation of the GluN2C subunit. Thus, 2i represents a robust tool for the GluN2C/D target validation. Esterification of the succinate carboxylate improved brain penetration, suggesting a strategy for therapeutic development of this series for NMDAR-associated neurological conditions.[1]
|
| 分子式 |
C28H19CL2F2N3O4
|
|---|---|
| 分子量 |
570.4
|
| 精确质量 |
569.0720678
|
| CAS号 |
2377187-09-0
|
| 外观&性状 |
Typically exists as Off-white to light yellow solid at room temperature
|
| LogP |
4.9
|
| tPSA |
99.1Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
39
|
| 分子复杂度/Complexity |
1050
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C1C(N(N=C1C2=C(C3=CC=CC=C3NC2=O)C4=CC=C(C=C4)Cl)C(=O)C(CC(=O)O)(F)F)C5=CC=C(C=C5)Cl
|
| InChi Key |
XJZPJUOPCITDPJ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C28H19Cl2F2N3O4/c29-17-9-5-15(6-10-17)22-13-21(34-35(22)27(39)28(31,32)14-23(36)37)25-24(16-7-11-18(30)12-8-16)19-3-1-2-4-20(19)33-26(25)38/h1-12,22H,13-14H2,(H,33,38)(H,36,37)
|
| 化学名 |
4-[3-(4-chlorophenyl)-5-[4-(4-chlorophenyl)-2-oxo-1H-quinolin-3-yl]-3,4-dihydropyrazol-2-yl]-3,3-difluoro-4-oxobutanoic acid
|
| 别名 |
DQP-997-74; 2377187-09-0; SCHEMBL16339958; XJZPJUOPCITDPJ-UHFFFAOYSA-N; 4-[3-(4-chlorophenyl)-5-[4-(4-chlorophenyl)-2-oxo-1H-quinolin-3-yl]-3,4-dihydropyrazol-2-yl]-3,3-difluoro-4-oxobutanoic acid
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7532 mL | 8.7658 mL | 17.5316 mL | |
| 5 mM | 0.3506 mL | 1.7532 mL | 3.5063 mL | |
| 10 mM | 0.1753 mL | 0.8766 mL | 1.7532 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Lalistat 2
Lalistat 2
 D-(-)-3-Phosphoglyceric acid disodium (3-Phospho-D-glyceric acid disodium)
D-(-)-3-Phosphoglyceric acid disodium (3-Phospho-D-glyceric acid disodium)
 STC-15
STC-15
 TSR-033
TSR-033
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























