| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
Fibroblast activation protein-α (FAP)
|
|---|---|
| 体外研究 (In Vitro) |
FAP抑制剂对人重组FAP的效力和选择性[1]
PNT6555、PNT6952和PNT6522在未与金属螯合时对FAP的IC50值较低,分别比PREP和DPP4的IC50值低200至1000倍和10000至30000倍(表1)。金属的引入使FAP的效力降低了2至48倍,具体取决于化合物(表1)。金属螯合也使PREP的亲和力降低了≤4倍。未检测到DPP4的抑制作用。 FAP抑制剂的细胞摄取和内化[1] 通过比较HEK mFAP和HEK Mock细胞的总摄取量,研究了细胞摄取natLu-PNT6555对FAP的依赖性。ICP-MS对总细胞相关natLu-PNT6555的测量表明,摄取需要FAP的表达(图2A)。1小时后HEK-mFAP细胞中natLu的总细胞结合和内化量的ICP-MS测量 h与natLu-PNT6555、natLu-PNT4952或natLu-PNT6522一起孵育表明natLu-PNT 6555表现出最大程度的内化(图2B)。 |
| 体内研究 (In Vivo) |
PNT6555、PNT6952和PNT6522 177Lu放射性配体的抗肿瘤活性[1]
177Lu-PNT6555、177Lu-PNT4952和177Lu-PNT 6522以15、30或60 MBq的单剂量给药于HEK-mFAP肿瘤携带小鼠。选择这些剂量是为了能够直接与之前在HEK-FAP肿瘤模型中评估的177Lu-FAPI-46和177Lu-FAP-2286进行疗效比较。177Lu放射性配体在肿瘤生长中产生剂量依赖性延迟,而未标记的前体没有产生明显的影响(图5A)。在对照组死亡前的肿瘤测量的最后一天,所有研究剂量的肿瘤生长都受到了显著抑制(补充图4A)。177Lu-PNT6555产生了最长的肿瘤生长延迟(图5A),疗效的等级顺序为177Lu-PNT 6555>177Lu-PNT4952>177Lu-NT6522。同样的等级顺序也反映在动物存活率上(图5B)。放射性配体似乎具有良好的耐受性,体重减轻≤10%且是短暂的(补充图5)。
225Ac-PNT6555和161Tb-PNT6555的抗肿瘤活性[1] 对于携带HEK-mFAP肿瘤的小鼠,225Ac-PNT6555以5、25或50 kBq的单次剂量给药,161Tb-PNT655以15、30或60 MBq的单剂量给药。225Ac-PNT6555剂量基于在PANC-1异种移植物模型中安全有效的单剂量225Ac-FAPI-46,161Tb-PNT655剂量基于安全有效的针对L1细胞粘附蛋白或叶酸受体的单剂量放射性金属。225Ac-PNT655和161Tb-PNT6555在所有剂量水平下都会导致肿瘤生长的剂量依赖性延迟(图6A),在对照动物死亡之前,肿瘤体积显著减少(补充图4B)。在225Ac-PNT6555实验中,接受非放射性标记PNT6555的小鼠的平均肿瘤体积似乎增加到高于载体治疗的小鼠。尽管这种效应在统计学上具有显著性,但它在药理学上没有意义,因为它很小,在177Lu-PNT6555(图5A)和161Tb-PNT655实验(图6A)中没有发现。225Ac-PNT655和161Tb-PNT6555以剂量依赖的方式增加了动物的存活率(图6B)。这两种放射性配体都具有良好的耐受性,对体重的影响很小(补充图6)。 |
| 酶活实验 |
体外荧光IC50测量[1]
测定了FAP配体对重组人FAP、DPP4和PREP的半最大抑制浓度(IC50)值。在室温下孵育FAP(pH 7.5)、PREP(pH7.5)和DPP4(pH 8.0)10分钟 在96孔板中用1:10的FAP配体(PNT6555、PNT6952和PNT6522)连续稀释。将7-氨基-4-甲基香豆素(AMC)荧光底物(羧基苄基(Z)-Gly-Pro-AMC用于FAP,PREP和Gly-Pro-AMC用于DPP4)以25µM的终浓度加入反应中。进一步孵育15天后 通过荧光法(Ex380nm:Em460nm)测量酶活性。以N-(4-喹啉酰基)-d-Ala-Pro(3144)-AMC为FAP底物的改良方法也用于重组人和小鼠FAP的IC50测定。酶在37°C下孵育10 用FAP配体的1:10系列稀释液稀释。加入3144-AMC,终浓度为25 μM,并继续孵育30分钟 荧光测定前,在37°C下至少进行一次。 对于血清样本的IC50测定,将1:10稀释的人血清和Sprague-Dawley大鼠血清以及1:100稀释的小鼠血清在37°C下孵育10分钟 用FAP配体的1:10系列稀释液稀释。加入3144-AMC,终浓度为25 μM(大鼠血清)或50 μM(人血清和小鼠血清),并继续孵育30分钟 荧光测定前,在37°C下至少进行一次。 对于细胞膜FAP的IC50测定,从生长至约80%融合的大块培养物中收获HEK-mFAP细胞,并将其铺在96孔板上。孵育过夜后,将1:10系列稀释的FAP配体与细胞孵育1小时 h.以20μg/ml的终浓度加入3144-AMC μM和37°C下继续孵育30分钟 荧光分析前分钟。 ICP-MS检测天然Lu螯合FAP配体的体外吸收和内化[1] 通过电感耦合等离子体质谱(ICP-MS)分析与HEK mFAP或HEK Mock细胞相关的总natLu,研究了natLu FAP配体的体外细胞摄取。内化的部分代表了酸洗去除任何细胞膜结合抑制剂后剩余的natLu。将HEK mFAP或HEK模拟细胞以4×106个细胞/2的比例接种在6孔板(Costar)中 mL/孔,在无血清RPMI 1640测定培养基中孵育(37°C,5%CO2)18-24小时 h.吸出培养基,用1、5、10或100替换 nM natLu-PNT6555用于测量FAP配体摄取或10 nM natLu-PNT6555、natLu-PNT4952或natLu-PNT 6522用于测量FAP配体的摄取和内化。孵育1小时后 在37°C下加热h,用1 mL冰冷的磷酸盐缓冲盐水(PBS)。为了测量细胞FAP配体的总摄取量(natLutotal),通过在室温下在0.3℃下孵育来裂解细胞 M氢氧化钠5 然后将样品通过23号针剪切DNA。对于细胞内化的测量,1 50毫升冰 mM甘氨酸-100 在上述PBS洗涤步骤后,将mM NaCl(pH 2.8)缓冲液加入细胞中。在4°C下孵育10天后 min,用1洗涤细胞两次 mL冰冷的PBS,然后如前所述裂解,以提供用于测量内化FAP配体(天然干扰素)的样品。通过Bradford测定法(Bio-Rad)测量总摄取和内化样品的蛋白质浓度。裂解后样品(300μL)用0.5 mL超纯水和2.0 mL超高纯硝酸。用超纯水进一步稀释样品,以获得2.8%的硝酸。内标(铟的最终浓度为5 ppb)添加到样品中并将0.1ppt的铟添加到500ppt的Lu标准中以创建标准曲线。ICP-MS以低分辨率模式进行。校准剂强度被归一化为内部对照的强度,并减去空白样品中的强度以创建线性校准曲线。HEK mFAP细胞与1、5、10或100μg/ml孵育后natLu的总细胞摄取量 nM natLu-PNT6555表示为绝对量(纳克)。natLu的内化百分比计算为(natLuinnal/natLutotal)×100。 |
| 动物实验 |
HEK-mFAP Mouse Tumor Xenograft Model [1]
Six-week-old male Fox Chase mice with severe combined immunodeficiency were injected subcutaneously with HEK-mFAP cells. Tumor growth was determined by measurement of tumor width (W) and length (L) with calipers, and tumor volume (V) expressed in mm3 was calculated by the formula V = (W2 × 0.5L). Formulation and In Vivo Administration of Radioligands [1] Radioligands diluted in PBS were administered to anesthetized mice by a single injection into the lateral tail vein. Biodistribution of 68Ga- and 177Lu-Radioligands In Vivo [1] HEK-mFAP tumor–bearing mice were injected intravenously with defined doses of 68Ga-PNT6555 or 177Lu-radioligands. At designated time points, blood and tissues were collected from 3 mice per treatment and counted for radioactivity. Tissue weights were measured for determination of the percentage injected dose per gram (%ID/g). PET and SPECT [1] After intravenous injection of radioligands, 68Ga imaging by small-animal PET/CT and 177Lu SPECT imaging were performed. Antitumor Activity of 177Lu-, 225Ac-, and 161Tb-Radioligands In Vivo [1] Mice bearing HEK-mFAP tumors of the volumes specified in the Results section were administered 177Lu-, 225Ac-, or 161Tb-radioligands, vehicle, or precursor ligands (6 mice per group) on day 1. Health checks were performed, and body weights and tumor dimensions were measured weekly. Mortality or euthanasia was used interchangeably for plotting mouse survival curves. Tumor growth curves were plotted up to the time of the earliest incidence of a mortality/euthanasia endpoint in each of the control and test groups. |
| 药代性质 (ADME/PK) |
Biodistribution of Radiolabeled FAP Inhibitors in Tumor-Bearing Mice [1]
68Ga-PNT6555 and 68Ga-PNT6952 exhibited selective uptake into HEK-mFAP tumors (Fig. 3A). Small-animal PET indicated that the radioligands rapidly entered the tumors, and while they were progressively cleared from the blood and normal tissues via the kidneys and the bladder, tumor activity increased rapidly over the first 5 min after injection, and more slowly but continuously thereafter (Fig. 3C; Supplemental Fig. 2). Elimination from blood, liver, and muscle resulted in radioligand levels that were distinctly lower in normal tissues than in tumors by ∼25 min after administration (Fig. 3C), and high-contrast PET images of tumors were obtained at 60 min (Fig. 3C). All 3 177Lu-FAP radioligands exhibited selective uptake in tumors 4 h after a single dose in HEK-mFAP tumor–bearing mice (Fig. 4A; Supplemental Fig. 3). Intratumoral levels of all 3 radioligands decreased between 4 and 48 h, but high tumor-to-normal tissue ratios were maintained from 48 to 168 h. Analysis of the area under the curve (AUC) ((%ID/g)·h) for the period from 4 to 168 h indicated that the accumulation of 177Lu-PNT6555 in the tumor was significantly greater than that for 177Lu-PNT6952 or 177Lu-PNT6522 (P < 0.0001) (Table 3; Supplemental Table 1), but the increase in 177Lu-PNT6555 between 48 and 168 h was not statistically significant. Although the limited uptake in normal tissues exhibited some variation between the 3 radioligands (Table 3), these differences were not statistically significant (Supplemental Table 1). The highest levels of uptake into normal tissues occurred in kidney, liver, bone and skin for 177Lu-PNT6555, kidney for 177Lu-PNT6952, and kidney and bone for 177Lu-PNT6522 (Table 3; Supplemental Fig. 3). However, the high initial tumor uptake and kinetics of tumor retention resulted in tumor-to-normal tissue AUC ratios of 15 to 19 in these tissues (Table 3). This is illustrated by the pharmacokinetic profiles in tumor and kidney (Fig. 4A), and the retention of 177Lu-PNT6555 in the tumors was apparent in SPECT images collected from 3 to 120 h (Fig. 4B). |
| 参考文献 | |
| 其他信息 |
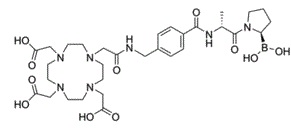
The overexpression of fibroblast activation protein-α (FAP) in solid cancers relative to levels in normal tissues has led to its recognition as a target for delivering agents directly to tumors. Radiolabeled quinoline-based FAP ligands have established clinical feasibility for tumor imaging, but their therapeutic potential is limited due to suboptimal tumor retention, which has prompted the search for alternative pharmacophores. One such pharmacophore is the boronic acid derivative N-(pyridine-4-carbonyl)-d-Ala-boroPro, a potent and selective FAP inhibitor (FAPI). In this study, the diagnostic and therapeutic (theranostic) potential of N-(pyridine-4-carbonyl)-d-Ala-boroPro-based metal-chelating DOTA-FAPIs was evaluated. Methods: Three DOTA-FAPIs, PNT6555, PNT6952, and PNT6522, were synthesized and characterized with respect to potency and selectivity toward soluble and cell membrane FAP; cellular uptake of the Lu-chelated analogs; biodistribution and pharmacokinetics in mice xenografted with human embryonic kidney cell-derived tumors expressing mouse FAP; the diagnostic potential of 68Ga-chelated DOTA-FAPIs by direct organ assay and small-animal PET; the antitumor activity of 177Lu-, 225Ac-, or 161Tb-chelated analogs using human embryonic kidney cell-derived tumors expressing mouse FAP; and the tumor-selective delivery of 177Lu-chelated DOTA-FAPIs via direct organ assay and SPECT. Results: DOTA-FAPIs and their natGa and natLu chelates exhibited potent inhibition of human and mouse sources of FAP and greatly reduced activity toward closely related prolyl endopeptidase and dipeptidyl peptidase 4. 68Ga-PNT6555 and 68Ga-PNT6952 showed rapid renal clearance and continuous accumulation in tumors, resulting in tumor-selective exposure at 60 min after administration. 177Lu-PNT6555 was distinguished from 177Lu-PNT6952 and 177Lu-PNT6522 by significantly higher tumor accumulation over 168 h. In therapeutic studies, all 3 177Lu-DOTA-FAPIs exhibited significant antitumor activity at well-tolerated doses, with 177Lu-PNT6555 producing the greatest tumor growth delay and animal survival. 225Ac-PNT6555 and 161Tb-PNT6555 were similarly efficacious, producing 80% and 100% survival at optimal doses, respectively. Conclusion: PNT6555 has potential for clinical translation as a theranostic agent in FAP-positive cancer. [1]
The selective inhibition of FAP over the dipeptidyl peptidases and PREP by 3099, which was achieved by the pyridin-4-carbonyl blocking group at the N terminus, and d-alanine at P2, respectively (25), was maintained in PNT6555, PNT6952, and PNT6522. When expressed as a cell membrane protein in HEK-mFAP cells, the catalytic site of FAP was found to be pharmacologically accessible to the d-Ala-boroPro–based ligands and to be essential for the cellular uptake of natLu-PNT6555 by HEK-mFAP cells. In vivo, 68Ga-PNT6555 and 68Ga-PNT6952 were selectively retained in HEK-mFAP tumors, resulting in PET images with high tumor-to-background contrast. The biodistribution of 177Lu-PNT6555, 177Lu-PNT6952, and 177Lu-PNT6522 in HEK-mFAP tumor–bearing mice confirmed the selective targeting of the HEK-mFAP tumors and revealed that 177Lu-PNT6555 exhibited the greatest tumor accumulation, consistent with its greater cellular internalization compared with natLu-PNT6952 and natLu-PNT6522 in vitro.[1] |
| 分子式 |
C31H48BN7O11
|
|---|---|
| 分子量 |
C31H48BN7O11
|
| 精确质量 |
705.35
|
| 元素分析 |
C, 52.77; H, 6.86; B, 1.53; N, 13.90; O, 24.94
|
| CAS号 |
2715113-34-9
|
| PubChem CID |
168489413
|
| 外观&性状 |
Typically exists as White to off-white solid at room temperature
|
| tPSA |
244 Ų
|
| 氢键供体(HBD)数目 |
7
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
14
|
| 重原子数目 |
50
|
| 分子复杂度/Complexity |
1140
|
| 定义原子立体中心数目 |
2
|
| SMILES |
B([C@@H]1CCCN1C(=O)[C@@H](C)NC(=O)C2=CC=C(C=C2)CNC(=O)CN3CCN(CCN(CCN(CC3)CC(=O)O)CC(=O)O)CC(=O)O)(O)O
|
| InChi Key |
PTVMRWQEZPTMFA-RDGATRHJSA-N
|
| InChi Code |
InChI=1S/C31H48BN7O11/c1-22(31(48)39-8-2-3-25(39)32(49)50)34-30(47)24-6-4-23(5-7-24)17-33-26(40)18-35-9-11-36(19-27(41)42)13-15-38(21-29(45)46)16-14-37(12-10-35)20-28(43)44/h4-7,22,25,49-50H,2-3,8-21H2,1H3,(H,33,40)(H,34,47)(H,41,42)(H,43,44)(H,45,46)/t22-,25+/m1/s1
|
| 化学名 |
2-[4-[2-[[4-[[(2R)-1-[(2R)-2-boronopyrrolidin-1-yl]-1-oxopropan-2-yl]carbamoyl]phenyl]methylamino]-2-oxoethyl]-7,10-bis(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetic acid
|
| 别名 |
BDBM609722; DOTA- aminomethyl-Bz- D-Ala-boroPro; US11707539, Compound 6555; 2715113-34-9
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO (~250 mg/mL (354 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
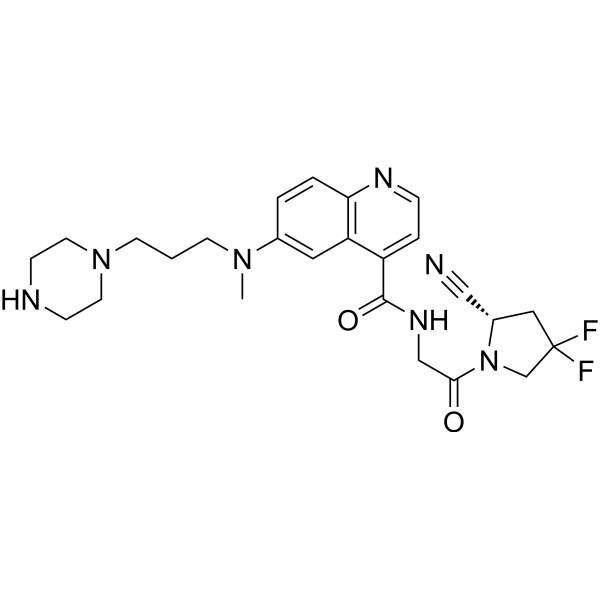 FT-FAPI-12_9
FT-FAPI-12_9
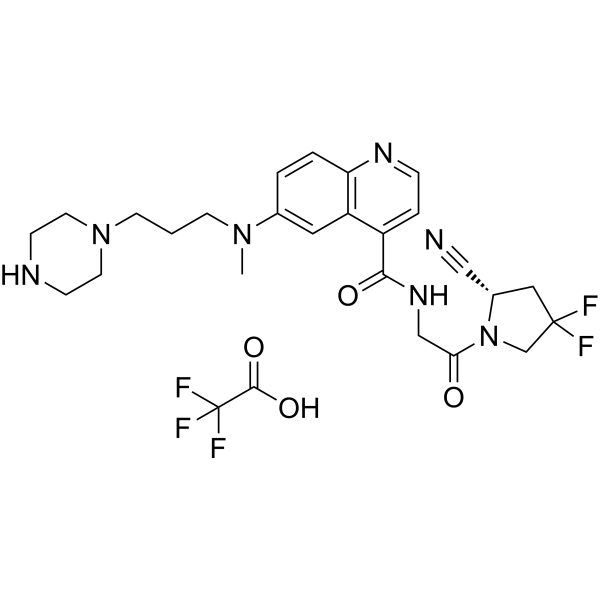 FT-FAPI-12_9 TFA
FT-FAPI-12_9 TFA
 FAPI-mFS
FAPI-mFS
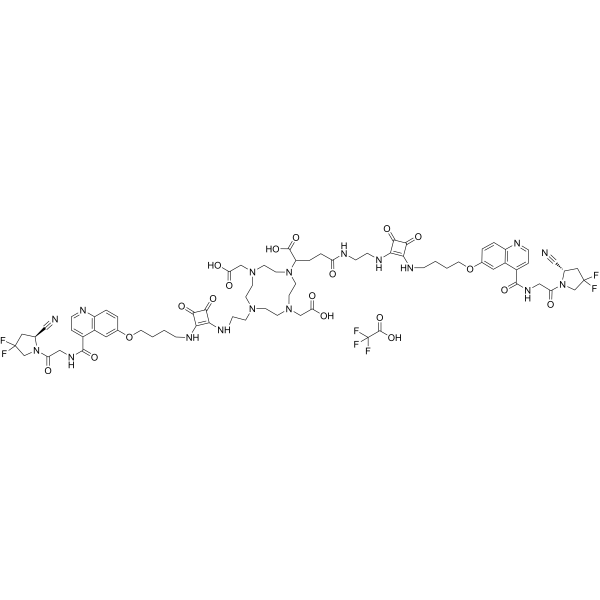 DOTAGA.(SA.FAPi)2 TFA
DOTAGA.(SA.FAPi)2 TFA
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
