| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
D1/dopamine receptor
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
(R)-SKF 38393。AtT-20 细胞、生长抑素或 D3 多巴胺受体产生的内源 GIRK 电流被盐酸 ((±)-SKF-38393;0-100 µM) 阻断,IC50 为 268 nM[5]。体外活性:SKF38393 (50-100 μM) 以蛋白质合成依赖性方式诱导持久的突触增强。在体外大鼠前额皮质神经元中,SKF 38393 模拟 DA 对 I(NaP) 的影响,并调节持续的钠电流。在听觉皮层中,SKF38393 通过激活下游效应器腺苷酸环化酶和磷脂酶 C 显着的蛋白质组改变来影响长期记忆的形成和巩固。激酶测定:SKF 38393 salthalide 是一种 D1 激动剂,IC50 为 110 nM。
儿茶酚胺、去甲肾上腺素和多巴胺(DA)存在于人类卵巢中;特别是在卵泡液中。去甲肾上腺素激活卵巢α和β肾上腺素受体并调节卵巢类固醇生成,但卵巢DA的意义尚不清楚。我们检查了D1亚型(D1-R)的DA受体是否存在于人类卵巢和培养的人类颗粒黄体细胞(GC)中。使用RT-PCR,我们从成人卵巢和GC信使RNA中克隆了与人类D1-R序列相同的互补DNA。在卵巢切片中,在大窦卵泡的颗粒细胞、黄体细胞以及培养的GC中鉴定出D1-R蛋白(通过免疫组织化学)。在Western印迹中使用相同的抗血清在培养的黄素化GC中发现了约50000 Mr的免疫反应带。这些细胞中的D1-R是功能性的,因为DA单独或在β受体拮抗剂普萘洛尔存在的情况下会引起细胞收缩。选择性D1-R激动剂SKF-38393诱导了细胞形态的类似变化,并增加了培养基cAMP的水平。然而,SKF-38393未能在体外显著影响基础和hCG刺激的孕酮释放,表明D1-R的激活与人类GC的主要类固醇孕酮的合成没有直接联系。雌二醇的合成同样不受影响。使用RT-PCR和免疫组织化学,我们发现GC表达DA和cAMP调节的Mr 32000磷酸化蛋白(DARPP-32),这是一种通常与携带D1-R的神经元相关的蛋白。综上所述,DA和功能性DA受体以及DARPP-32的存在表明,人类卵巢中存在一种涉及DA的新的生理调节途径[2]。 |
||
| 体内研究 (In Vivo) |
SKF 38393(6 mg/kg,腹腔注射)可防止东莨菪碱引起的 T 迷宫工作记忆任务表现受损。在成年雄性 NMRI 小鼠中,SKF38393(1 μg/小鼠)会损害情境依赖性恐惧学习。
在这项研究中,研究人员检查了多巴胺能(D1)受体激动剂SKF-38393 HCl(SKF)对MPTP诱导的多巴胺能神经元损伤的可能神经保护作用。MPTP被单胺氧化酶-B(MAO-B)转化为其神经毒性代谢产物1-甲基-4-苯基吡啶鎓(MPP+),然后被多巴胺能神经元吸收。SKF-38393对纹状体中的总氧化酶或单胺氧化酶B均无影响。SKF-38393阻断了MPTP诱导的谷胱甘肽耗竭,并减轻了MPTP引起的多巴胺耗竭。此外,它增强了超氧化物歧化酶的活性,从而模拟了司来吉兰的作用。这些研究的结果被解释为表明,SKF-38393可能被证明是治疗帕金森病的一种有价值的药物。[3] |
||
| 酶活实验 |
SKF 38393 saltloride 是 D1 的激动剂,IC50 为 110 nM。
使用定量放射自显影检查碘化SCH 23390、125I-SCH 23982(杜邦NEN)在大鼠脑切片中与多巴胺D1受体结合的效力、选择性以及解剖和神经元定位。125I-SCH 23982以非常高的亲和力(Kd值为55-125pM)、特异性(70-85%的结合被5微摩尔顺式氟戊噻醇取代)和可饱和的方式(Bmax值为65-176fmol/mg蛋白)结合基底节中的D1位点。选择性D1拮抗剂SCH 23390(IC50=90 pM)和顺式氟戊噻醇(IC50=200 pM)以及D1激动剂SKF 38393(IC50=110 nM)取代了特异性125I-SCH 23982结合,但D2选择性配体(I-舒必利,LY 171555)或S2拮抗剂西那塞林没有取代。与3H-SCH 23390相比,125I-SCH 23882对D1位点的亲和力提高了5到10倍,比放射性提高了50倍,使其成为标记D1受体的优秀放射性配体。D1位点的浓度在内侧黑质中最高,超过外侧黑质、尾壳核、伏隔核、嗅结节和内脚核中D1位点浓度的50%以上。较低浓度的D1位点存在于内囊、背内侧额叶皮层、屏状核和新皮层第6层。腹侧被盖区缺失D1位点。纹状体注射保留轴突的神经毒素喹啉酸,分别使同侧尾壳核和黑质内侧和中央网状部可移位D1位点的浓度减少87%和46-58%。黑质外侧未见D1位点丢失。用6-羟基多巴胺破坏高达94%的中脑多巴胺能投射并没有减少D1结合,也没有增加纹状体或黑质D1受体浓度,只有一个例外。125I-SCH 23982以皮摩尔亲和力选择性标记纹状体神经元上的D1结合位点,这些神经元包含大鼠大脑中的大部分D1位点[1]。 |
||
| 细胞实验 |
细胞系:GC细胞
浓度:10 μmol/L 孵育时间:1小时 结果:诱导Mr 32 kD (DARPP-32)的DA-和cAMP-调节的磷蛋白的苏氨酸磷酸化增加培养GC细胞。 蛋白质印迹[2] 如前所述进行蛋白质印迹,但稍作修改。简而言之,收集细胞,冷冻、解冻,在含有10%蔗糖和2%SDS的62.5 mmol/L Tris-HCl缓冲液(pH 6.8)中均质化,超声处理,并在10%巯基乙醇存在下加热(95℃5分钟)。样品(15μg/泳道)在10%或12.5%SDS-聚丙烯酰胺凝胶(SDS-PAGE)上进行电泳分离。将蛋白质转移到硝化纤维膜上,并用用于免疫组织化学的相同D1-R抗血清(1:1000稀释,在4℃下孵育过夜)进行检测。 此外,使用特征明确的单克隆磷酸-DARPP-32特异性抗体(1:500)检测用DA(1和10μmol/L)或SKF-38393(1和1μmol/L,RBI,Biotrend,Cologne,Germany,稀释于无血清培养基中)处理GC(1小时,2例,24小时)是否改变了DARPP-32的磷酸化。为了控制目的,将β受体拮抗剂普萘洛尔(10μmol/L)或D1-R拮抗剂SCH-23390(10μmol/L,RBI)添加到用DA或SKF-38393(在1μm处使用)处理的细胞中。监测并记录细胞形态。如所述,使用过氧化物酶标记的抗血清(1:3000)和增强化学发光检测免疫反应性。在某些情况下,印迹被数字化,并使用NIH Image程序的编辑版本确定条带的积分光密度,如前所述。 免疫沉淀实验[2] 进行免疫沉淀实验,以检查SKF-38393(100μmol/L,在无血清培养基中稀释)处理培养的GC(分离后1天)1小时是否会增加DARPP-32的苏氨酸磷酸化。移除培养基,将细胞溶解在含有10 mmol/L NaH2PO4、150 mmol/L NaCl、2 mmol/L EDTA、1%Triton X-100、0.25%SDS、1%脱氧胆酸钠和2 mmol/L苯基甲磺酰氟的缓冲液中。为了进行免疫沉淀,我们使用了标记有抗鼠IgG的磁珠和磁分离。首先将珠子与正常小鼠血清(5%在含有10 mmol/L EGTA、250 mmol/L蔗糖和0.1%BSA的PBS中)一起孵育,然后用2μL特征明确的针对牛DARPP-32的单克隆抗体标记,该抗体可识别灵长类动物DARPP-32,也用于免疫细胞化学。随后,将珠子与150μL GC细胞裂解液在室温下孵育1小时,然后在4℃下孵育30分钟。磁铁分离后,将小球洗涤几次并用于SDS-PAGE,如所述。使用抗磷酸苏氨酸的单克隆抗体(1:100)进行印迹;在某些情况下,它们是通过密度测定法进行评估的。 孕酮和雌二醇测量[2] 在hCG(10IU/mL)存在或不存在的情况下,通过GC与SKF-38393(10μmol/L)一起孵育6小时,使用三个孔对每种处理(n=3)检测孕酮和雌二醇在培养基中的释放情况。按照制造商的说明,使用商业酶免疫测定法对样品进行分析。批内变异系数在5-8%之间,批间变异系数不超过10%。所有孵育和移液步骤以及激素浓度的计算都在全自动免疫诊断分析仪中进行。对细胞蛋白的微小变化进行了校正。方差分析和t检验用于评估结果。 cAMP的测定[2] 在磷酸二酯酶抑制剂异丁甲基黄嘌呤(1 mmol/L)的存在下,用SKF-38393(1-100μmol/L)孵育3或6小时后,检测分离后1天GC培养基中cAMP的水平。在一项初步研究中,SKF-38393(浓度为1μmol/L)导致cAMP小幅但无统计学意义的增加(比基础水平高20%)。因此,对于三个独立的附加实验,使用了更高的SKF-38393浓度(100μmol/L)。根据制造商的说明,这些样品是使用酶免疫测定法(R&D Systems)测量的。该测定的灵敏度为0.5 pmol/mL,批内变异系数小于10%。为了校正细胞密度的微小差异,每微克细胞蛋白表达cAMP结果。学生t检验用于评估数据。 R-(+)-7-氯-8-羟基-3-甲基-1-苯基-2,3,4-5-四氢-1H-3-苯扎氮平盐酸盐(SCH23390)是一种广泛使用的D1多巴胺受体高选择性拮抗剂。在研究D1和D3多巴胺受体信号通路之间的串扰时,我们发现,除了作为D1受体拮抗剂外,SCH23390和相关化合物还抑制G蛋白偶联内向整流钾(GIRK)通道。我们提出的证据表明,SCH23390阻断了AtT-20细胞中生长抑素或D3多巴胺受体诱导的内源性GIRK电流(IC50,268 nM)。这种抑制是受体非依赖性的,因为仅表达GIRK通道的中国仓鼠卵巢细胞中的组成型GIRK电流也被SCH23390阻断。GIRK通道的抑制具有一定的选择性,因为与内向整流钾通道密切相关的Kir2.0家族成员以及AtT-20细胞中存在的各种内源性阳离子电流不受影响。此外,在电流钳记录中,SCH23390可以使膜电位去极化,并诱导AtT-20细胞释放动作电位,表明GIRK通道抑制的潜在生理意义。为了确定导致GIRK通道阻断的化学特征,我们测试了几种结构相关的化合物[R(+)-SKF-38393A,R-(+)-7-氯-8-羟基-1-苯基-2,3,4-5-四氢-1H-3-苯并氮杂卓盐酸盐(nor-methyl-SCH23390)和R-(+”)-2,3,4,5-四氢-8-碘-3-甲基-5-苯基-1H-3-苯并咪唑-7-醇盐酸盐(iodo-SCH23390)],我们的结果表明卤化物原子对阻断GIRK信道至关重要。综上所述,我们的结果表明,SCH23390和相关化合物可能为设计新型GIRK通道选择性阻断剂提供依据。也许更重要的是,一些专门使用SCH23390来探测D1受体功能或作为D1受体参与诊断的研究可能需要根据这些结果重新评估[5]。 |
||
| 动物实验 |
|
||
| 参考文献 |
|
||
| 其他信息 |
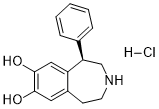
R-(+)-7-氯-8-羟基-3-甲基-1-苯基-2,3,4,5-四氢-1H-3-苯并氮杂卓盐酸盐 (SCH23390) 是一种广泛使用的、高选择性的D1多巴胺受体拮抗剂。在研究D1和D3多巴胺受体信号通路之间的相互作用时,我们发现SCH23390及其相关化合物除了作为D1受体拮抗剂外,还能抑制G蛋白偶联的内向整流钾(GIRK)通道。我们提供的证据表明,SCH23390能够阻断AtT-20细胞中由生长抑素或D3多巴胺受体诱导的内源性GIRK电流(IC50为268 nM)。这种抑制作用与受体无关,因为在仅表达GIRK通道的中国仓鼠卵巢细胞中,组成型GIRK电流也被SCH23390阻断。 GIRK通道的抑制具有一定的选择性,因为与其密切相关的Kir2.0家族内向整流钾通道以及AtT-20细胞中存在的各种内源性阳离子电流均不受影响。此外,在电流钳记录中,SCH23390能够使膜电位去极化并诱发AtT-20细胞产生动作电位,这表明GIRK通道抑制可能具有生理意义。为了确定导致GIRK通道阻滞的化学特征,我们测试了几种结构相关的化合物[R(+)-SKF-38393A、R-(+)-7-氯-8-羟基-1-苯基-2,3,4,5-四氢-1H-3-苯并氮杂卓盐酸盐(nor-methyl-SCH23390)和R-(+)-2,3,4,5-四氢-8-碘-3-甲基-5-苯基-1H-3-苯并氮杂卓-7-醇盐酸盐(iodo-SCH23390)],结果表明卤素原子对于GIRK通道的阻滞至关重要。综上所述,我们的研究结果提示SCH23390及其相关化合物可能为设计新型GIRK通道选择性阻滞剂提供基础。或许更重要的是,一些专门使用 SCH23390 来探究 D1 受体功能或作为 D1 受体参与诊断的研究可能需要根据这些结果重新评估[1]。
一种选择性 D1 多巴胺受体激动剂,主要用作研究工具。 使用定量放射自显影技术检测了碘化 SCH 23390,125I-SCH 23982(杜邦-NEN),以研究其效力、选择性以及与大鼠脑切片中多巴胺 D1 受体结合的解剖学和神经元定位。 125I-SCH 23982 与基底神经节中的 D1 位点结合,具有非常高的亲和力(Kd 值为 55-125 pM)、特异性(5 μM 顺式氟哌噻吨可置换 70-85% 的结合),并且以可饱和的方式结合(Bmax 值为 65-176 fmol/mg 蛋白质)。选择性D1受体拮抗剂SCH 23390(IC50 = 90 pM)和顺式氟哌噻吨(IC50 = 200 pM)以及D1受体激动剂SKF-38393(IC50 = 110 nM)可取代125I-SCH 23982的特异性结合,但D2受体选择性配体(125I-舒必利、LY 171555)或S2受体拮抗剂西那色林则无此作用。与3H-SCH 23390相比,125I-SCH 23982对D1受体的亲和力高5~10倍,比放射性高50倍,使其成为标记D1受体的优良放射性配体。 D1受体的浓度在黑质内侧部最高,比黑质外侧部、尾状核壳核、伏隔核、嗅结节和内侧苍白球的浓度高出50%以上。内囊、背内侧额叶皮层、屏状核和新皮层第6层中D1受体的浓度较低。腹侧被盖区未检测到D1受体。纹状体内注射轴突保护性神经毒素喹啉酸后,同侧尾状核壳核和黑质网状部内侧部及中央部的可置换D1受体浓度分别降低了87%和46-58%。黑质外侧部的D1受体未见减少。用6-羟基多巴胺破坏高达94%的中脑纹状体多巴胺能投射,并未降低D1受体的结合,除一个例外,也未增加纹状体或黑质的D1受体浓度。125I-SCH 23982以皮摩尔级的亲和力选择性地标记纹状体黑质神经元上的D1受体结合位点,而这些神经元含有大鼠脑内大部分的D1受体位点。[1]帕金森病(PD)的特征是黑质纹状体多巴胺能神经元的进行性退化。线粒体呼吸抑制、羟自由基的产生以及导致氧化应激的自由基防御机制减弱等多种因素被认为与多巴胺能神经元的退化有关。 1-甲基-4-苯基-1,2,3,6-四氢吡啶 (MPTP) 处理的动物模型是帕金森病 (PD) 的有效实验模型,能够展现该疾病的大部分临床特征以及主要的生化和病理症状。本研究探讨了多巴胺能 (D1) 受体激动剂 SKF-38393 HCl (SKF) 对 MPTP 诱导的多巴胺能神经元损伤的潜在神经保护作用。MPTP 经单胺氧化酶 B (MAO-B) 转化为其神经毒性代谢物 1-甲基-4-苯基吡啶鎓 (MPP+),后者随后被多巴胺能神经元摄取。SKF-38393 对纹状体中的总 MAO-B 或 MAO-B 均无影响。 SKF-38393 阻断了 MPTP 诱导的谷胱甘肽耗竭,并减弱了 MPTP 诱导的多巴胺耗竭。此外,它还增强了超氧化物歧化酶的活性,从而模拟了司来吉兰的作用。这些研究结果表明,SKF-38393 可能是一种治疗帕金森病的有效药物。[3] 本研究旨在更好地评估多巴胺在胞吐作用中的作用。由于腺苷酸环化酶的直接激活(例如,使用福斯克林)可增强神经递质的释放,因此我们感兴趣的是,激活与腺苷酸环化酶正向偶联的 D1 型多巴胺受体是否也能调节突触小泡融合和神经递质释放的分子机制。为了回答这个问题,我们研究了D1型多巴胺受体激动剂SKF-38393对培养的大鼠海马神经元自发释放谷氨酸的影响。SKF-38393增强了河豚毒素耐受性兴奋性突触后电流的频率,但并未增强其幅度,这表明D1受体的作用位点位于突触前。D1多巴胺受体拮抗剂SCH-23390以及蛋白激酶A抑制剂H-7和Rp-cAMP均能阻断这种效应,而百日咳毒素则对多巴胺能反应没有影响。此外,卡巴胆碱和钌红也能刺激胞吐作用,但不能阻断SKF-38393诱导的调节作用。这些结果表明,SKF-38393 通过一种对百日咳毒素不敏感且依赖于蛋白激酶 A 的机制,在突触前增强谷氨酸的释放,该机制很可能涉及 D1 型多巴胺受体。我们的结果强调了蛋白激酶 A 作为突触传递的强效调节因子的重要性,并提示高浓度的多巴胺可以显著增强海马中谷氨酸的释放。[4] |
| 分子式 |
C16H17NO2.HCL
|
|
|---|---|---|
| 分子量 |
291.77
|
|
| 精确质量 |
291.103
|
|
| 元素分析 |
C, 65.86; H, 6.22; Cl, 12.15; N, 4.80; O, 10.97
|
|
| CAS号 |
81702-42-3
|
|
| 相关CAS号 |
SKF 38393 hydrochloride;62717-42-4; SKF 38393 hydrobromide; 20012-10-6; (R)-SKF 38393 hydrochloride; 81702-42-3; 62751-59-1 (R-isomer); 67287-49-4
|
|
| PubChem CID |
6852375
|
|
| 外观&性状 |
Typically exists as solid at room temperature
|
|
| LogP |
3.506
|
|
| tPSA |
52.49
|
|
| 氢键供体(HBD)数目 |
4
|
|
| 氢键受体(HBA)数目 |
3
|
|
| 可旋转键数目(RBC) |
1
|
|
| 重原子数目 |
20
|
|
| 分子复杂度/Complexity |
291
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
OC1C(O)=CC2=C([C@@H](C3C=CC=CC=3)CNCC2)C=1.Cl
|
|
| InChi Key |
YEWHJCLOUYPAOH-PFEQFJNWSA-N
|
|
| InChi Code |
InChI=1S/C16H17NO2.ClH/c18-15-8-12-6-7-17-10-14(13(12)9-16(15)19)11-4-2-1-3-5-11;/h1-5,8-9,14,17-19H,6-7,10H2;1H/t14-;/m1./s1
|
|
| 化学名 |
(5R)-5-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine-7,8-diol;hydrochloride
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.4274 mL | 17.1368 mL | 34.2736 mL | |
| 5 mM | 0.6855 mL | 3.4274 mL | 6.8547 mL | |
| 10 mM | 0.3427 mL | 1.7137 mL | 3.4274 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Proc Natl Acad Sci U S A. 1995 Mar 28; 92(7): 2446–2450. |
Proc Natl Acad Sci U S A. 1995 Mar 28; 92(7): 2446–2450. |
Proc Natl Acad Sci U S A. 1995 Mar 28; 92(7): 2446–2450. |
 艾洛司他
艾洛司他
 PROTAC-VHL-ligand
PROTAC-VHL-ligand
 ML132
ML132
 BAY-677
BAY-677
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
-SKF-38393A")
-SKF-38393A")
-SKF-38393A")
 463611831
463611831
