| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
Histamine H2 receptor
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:雷尼替丁使肝细胞对活化中性粒细胞的细胞毒性产物的杀伤敏感,而法莫替丁缺乏这种能力。雷尼替丁可抑制体外脂多糖刺激的单核细胞中肿瘤坏死因子-α (TNF-α) 的产生。雷尼替丁剂量依赖性地降低吗啡的 Kel,最大效果为 50%,并增加离体豚鼠肝细胞中 6-葡萄糖醛酸吗啡与 3-葡萄糖醛酸吗啡的相对浓度。雷尼替丁逐渐降低吗啡-3-葡萄糖醛酸/吗啡-6-葡萄糖醛酸的比例,最高可达 21%。
|
| 体内研究 (In Vivo) |
雷尼替丁导致大鼠肝损伤,证据是大鼠服用雷尼替丁后 6 小时内血清丙氨酸转氨酶、天冬氨酸转氨酶和 γ-谷氨酰转移酶活性增加。雷尼替丁可抑制大鼠肝缺血/再灌注引起的肝组织 TNF-α、细胞因子诱导的中性粒细胞趋化剂水平的增加以及中性粒细胞的肝脏积聚。雷尼替丁联合治疗可增强 LPS 诱导的肝损伤前的凝血,而抗凝剂可减轻 LPS/RAN 治疗大鼠的肝损伤。雷尼替丁/LPS治疗的大鼠导致肝窦中纤维蛋白凝块的形成,并防止与减少肝细胞损伤相关的纤维蛋白沉积。雷尼替丁联合治疗可增强大鼠肝细胞损伤发生前 LPS 诱导的 TNF 增加。雷尼替丁在高架十字迷宫中显示出抗焦虑作用,这通过大鼠张开臂的时间增加、更多的张开臂扫描和更多的末端偏移来表明。
|
| 细胞实验 |
在离体豚鼠肝细胞中研究雷尼替丁对吗啡代谢的影响,特别强调吗啡-3-葡糖苷和吗啡-6-葡糖苷的比值。雷尼替丁对吗啡的剂量依赖性最大可降低50%,使吗啡-6-葡糖苷的相对浓度增加到吗啡-3-葡糖苷的相对浓度。这些影响可能是由于对所涉及的偶联酶的直接或间接影响,或对吗啡或葡萄糖醛酸盐跨细胞膜运输的影响。后一种解释被拒绝,因为观察到吗啡、吗啡-3-葡糖苷和吗啡-6-葡糖苷细胞内和细胞外浓度的比值不受雷尼替丁的影响。雷尼替丁浓度的增加使吗啡-3-葡糖苷/吗啡-6-葡糖苷的比值逐渐降低了21%。这可能源于能量或共底物供应的干扰,或通过对不同UDPGTases的直接影响。观察到,目前对吗啡葡糖醛酸化的影响与给予已知的共底物(UDPGA)消耗物时所观察到的相反,表明雷尼替丁的作用很可能是直接抑制尿苷5'-二磷酸葡糖醛酸转移酶,对负责3'-葡糖醛酸化的同功酶的作用更为明显。[3]
|
| 动物实验 |
药物特异反应是指少数服用某种药物的人群中发生的病因不明的不良反应。某些特异反应可能由药物肝毒性阈值的间歇性降低引起。既往大鼠研究表明,由细菌脂多糖 (LPS) 诱发的轻度炎症可降低异源物质肝毒性阈值。组胺-2 (H2) 受体拮抗剂雷尼替丁 (RAN) 可引起人类特异反应,肝脏通常是其靶器官。研究人员检验了以下假设:在经历轻度炎症反应的动物中,RAN 可能具有肝毒性。[1]
雄性大鼠接受非肝毒性剂量的 LPS(44 x 10⁶ 内毒素单位/kg,静脉注射)或其溶剂,2 小时后接受非肝毒性剂量的 RAN(30 mg/kg,静脉注射)或其溶剂。仅在同时接受雷帕霉素(RAN)和脂多糖(LPS)治疗的动物中观察到肝损伤,表现为RAN给药后6小时内血清丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)和γ-谷氨酰转移酶(GGT)活性升高。LPS/RAN联合治疗导致肝脏中部出现病变,其特征为急性坏死性化脓性肝炎。法莫替丁(FAM)是一种H2受体拮抗剂,其特异性反应的发生率远低于RAN。给予大鼠与RAN药理效力相当的LPS和FAM后,未观察到肝损伤。体外实验表明,RAN可使肝细胞对活化中性粒细胞产生的细胞毒性产物更加敏感,而FAM则不具备这种能力。结果表明,在轻度炎症期间,动物暴露于雷尼替丁(RAN)可重现类似于人类雷尼替丁特异性反应的症状。[1] 此前有研究报道,H₂受体拮抗剂雷尼替丁可在体外和体内抑制中性粒细胞活化,从而有助于减轻大鼠应激诱导的胃黏膜损伤。本研究旨在探讨雷尼替丁是否能减轻大鼠缺血/再灌注引起的肝损伤(活化的中性粒细胞在其中起着关键作用)。此外,研究人员还考察了另一种H₂受体拮抗剂法莫替丁对体外和缺血/再灌注引起的肝损伤后大鼠白细胞活化的影响,以探究雷尼替丁对中性粒细胞活化的抑制作用是否依赖于其对H₂受体的阻断。如先前报道,雷尼替丁在体外抑制了中性粒细胞的活化,而法莫替丁则显著增强了中性粒细胞的活化。雷尼替丁可抑制脂多糖体外刺激的单核细胞中肿瘤坏死因子-α (TNF-α) 的产生,而法莫替丁则无此作用。尽管静脉注射30 mg/kg雷尼替丁可抑制肝脏缺血/再灌注引起的肝组织中TNF-α水平升高、细胞因子诱导的中性粒细胞趋化因子水平升高以及中性粒细胞在肝脏中的聚集,但静脉注射5 mg/kg法莫替丁却显著增强了这些升高。雷尼替丁可显著抑制再灌注后肝组织血流量和胆汁分泌的减少以及血清转氨酶水平的升高,而法莫替丁组动物的这些变化比对照组更为显著。这些观察结果强烈提示,雷尼替丁可通过直接抑制中性粒细胞活化,或通过抑制中性粒细胞强效活化剂TNF-α的产生而间接抑制中性粒细胞活化,从而减轻缺血/再灌注引起的肝损伤。此外,雷尼替丁的治疗效果可能并非仅仅由其对H(2)受体的阻断作用所致。[2] |
| 药代性质 (ADME/PK) |
代谢/代谢物
雷尼替丁已知的代谢物包括去甲基雷尼替丁。 |
| 毒性/毒理 (Toxicokinetics/TK) |
妊娠期和哺乳期影响
◉ 哺乳期用药概述 尽管个体间存在差异,但母乳中的雷尼替丁剂量低于新生儿用药剂量。然而,由于发现雷尼替丁会自发分解成致癌化学物质,因此在美国和其他国家/地区已将其撤出市场。建议使用其他药物。 ◉ 对母乳喂养婴儿的影响 一名54日龄的母乳喂养婴儿在母亲连续两天每12小时服用150毫克雷尼替丁后,未观察到任何不良反应。 ◉ 对泌乳和母乳的影响 已知组胺H2受体阻滞剂会刺激催乳素分泌。一些研究表明,静脉注射雷尼替丁超过100毫克或长期口服雷尼替丁会导致血清催乳素水平升高,并有罕见的男性乳房发育症病例报告。对于已建立泌乳的母亲而言,催乳素水平可能不会影响其哺乳能力。 |
| 参考文献 | |
| 其他信息 |
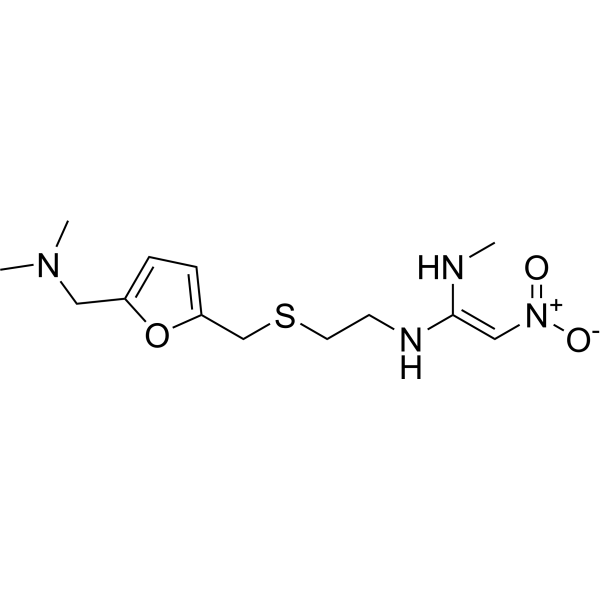
雷尼替丁属于呋喃类药物,用于治疗消化性溃疡和胃食管反流病。它具有抗溃疡药、H2受体拮抗剂、环境污染物、外源性物质和药物过敏原等多种作用。它属于呋喃类化合物、叔胺化合物、C-硝基化合物和有机硫化物。
雷尼替丁是具有抗酸活性的组胺H2受体拮抗剂。雷尼替丁是肠嗜铬样细胞(ECL细胞)释放的组胺与胃壁细胞上的组胺H2受体结合的竞争性可逆抑制剂,从而抑制胃酸的正常分泌和进食刺激引起的胃酸分泌。此外,当H2受体被阻断时,其他促进胃酸分泌的物质对壁细胞的作用也会减弱。 盐酸雷尼替丁属于组胺H2受体拮抗剂类抗酸剂。雷尼替丁是肠嗜铬样细胞(ECL细胞)释放的组胺与胃壁细胞上的组胺H2受体结合的竞争性可逆抑制剂,从而抑制胃酸的正常分泌和进食刺激引起的胃酸分泌。此外,当H2受体被阻断时,其他促进胃酸分泌的物质对壁细胞的作用也会减弱。 雷尼替丁是一种非咪唑类组胺受体(H2受体)阻断剂,该受体介导胃酸分泌。它用于治疗胃肠道溃疡。 另见:雷尼替丁(注释已移至)。 暴露于非毒性剂量的细菌脂多糖 (LPS) 会增加组胺-2 (H2) 受体拮抗剂雷尼替丁 (RAN) 的肝毒性。由于与 LPS 相关的一些病理生理效应是通过肿瘤坏死因子 α (TNF) 等炎症介质的表达和释放介导的,因此本研究旨在深入了解 TNF 在 LPS/RAN 肝毒性中的作用。为了确定雷帕霉素(RAN)是否影响肝损伤早期LPS诱导的TNF释放,我们用2.5 × 10⁶内毒素单位(EU)/kg LPS或其生理盐水(静脉注射)处理雄性Sprague-Dawley大鼠,2小时后,再用30 mg/kg RAN或无菌磷酸盐缓冲液(静脉注射)处理。LPS给药导致循环TNF浓度升高。RAN联合治疗增强了LPS诱导的TNF升高,且这种升高发生在肝细胞损伤发生之前,而法莫替丁(一种无特异性反应的H2受体拮抗剂)则没有这种作用。血清白细胞介素(IL)-1β、IL-6和IL-10也观察到类似的变化。为了确定TNF是否在LPS/RAN诱导的肝毒性中起因果作用,研究人员分别给予大鼠己酮可可碱(PTX;100 mg/kg,静脉注射)以抑制TNF的合成,或给予依那西普(Etan;8 mg/kg,皮下注射)以阻断TNF与细胞受体的结合,随后用LPS和RAN处理大鼠。研究人员评估了肝细胞损伤、炎症介质的释放、肝脏中性粒细胞(PMN)的聚集以及凝血和纤溶的生物标志物。结果显示,PTX或Etan预处理均可减轻LPS/RAN联合处理动物的肝损伤,并降低循环中TNF、IL-1β、IL-6、巨噬细胞炎症蛋白-2以及凝血/纤溶生物标志物的浓度。然而,PTX或Etan预处理均未改变肝脏PMN的聚集。这些结果表明,TNF通过增强炎症细胞因子的产生和止血作用,促进LPS/RAN诱导的肝损伤。[4] 本研究探讨了单侧注射H1受体拮抗剂氯苯那敏和H2受体拮抗剂雷尼替丁后,对基底核大细胞核(NBM)附近强化和焦虑参数的影响。在实验1中,将慢性植入导管的大鼠注射氯苯那敏或雷尼替丁(剂量分别为0.1、1、10和20 μg),然后将其置于圆形开放场(封闭围栏)的四个限制象限之一中进行一次条件反射训练。在条件性围栏偏好测试中,当大鼠在四个象限之间进行选择时,只有注射10或20 μg氯苯那敏的大鼠在治疗围栏中停留的时间更长,表明氯苯那敏具有正强化作用。其他剂量的氯苯那敏或H2受体拮抗剂均未影响大鼠的偏好行为。在实验2中,我们使用高架十字迷宫(EPM)来评估基底膜内注射氯苯那敏或雷尼替丁(剂量分别为0.1、1、10和20 μg)可能产生的抗焦虑或致焦虑作用。结果发现,单次注射0.1或20 μg的氯苯那敏以及20 μg的雷尼替丁均在EPM中表现出类似抗焦虑的作用。两种化合物均增加了大鼠在开放臂上的停留时间,并增加了其在开放臂边缘的扫描行为。其他剂量的H1和H2受体拮抗剂均未影响大鼠在EPM中的行为。总之,这些研究结果表明,H1 和 H2 受体拮抗剂对 NBM 中的强化和恐惧相关过程具有不同的调节作用,因此,首次证明了组胺能神经支配该脑区具有行为相关性。[5] |
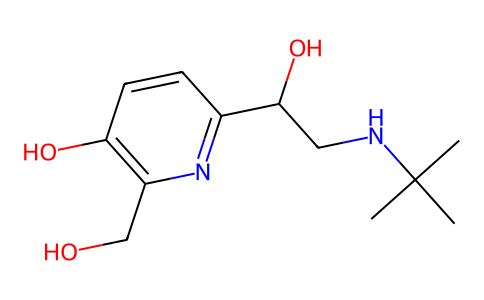
| 分子式 |
C13H22N4O3S.HCL
|
|---|---|
| 分子量 |
350.87
|
| 精确质量 |
314.141
|
| 元素分析 |
C, 49.66; H, 7.05; N, 17.82; O, 15.27; S, 10.20
|
| CAS号 |
66357-35-5
|
| 相关CAS号 |
Ranitidine hydrochloride;66357-59-3
|
| PubChem CID |
3001055
|
| 外观&性状 |
Off-white to light brown solid at room temperature
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
437.1±45.0 °C at 760 mmHg
|
| 熔点 |
69-70 °C
; MP: 133-134 °C /RATINIDINE HYDROCHLORIDE/
|
| 闪点 |
218.2±28.7 °C
|
| 蒸汽压 |
0.0±1.0 mmHg at 25°C
|
| 折射率 |
1.559
|
| LogP |
1.23
|
| tPSA |
111.56
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
21
|
| 分子复杂度/Complexity |
347
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CNC(=C[N+](=O)[O-])NCCSCC1=CC=C(CN(C)C)O1
|
| InChi Key |
VMXUWOKSQNHOCA-UKTHLTGXSA-N
|
| InChi Code |
InChI=1S/C13H22N4O3S/c1-14-13(9-17(18)19)15-6-7-21-10-12-5-4-11(20-12)8-16(2)3/h4-5,9,14-15H,6-8,10H2,1-3H3/b13-9+
|
| 化学名 |
(E)-1-N'-[2-[[5-[(dimethylamino)methyl]furan-2-yl]methylsulfanyl]ethyl]-1-N-methyl-2-nitroethene-1,1-diamine
|
| 别名 |
ranitidine; 66357-35-5; Ranitidine Base; ranitidine hydrochloride; Raticina; Coralen; Gastrial; Quantor;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8501 mL | 14.2503 mL | 28.5006 mL | |
| 5 mM | 0.5700 mL | 2.8501 mL | 5.7001 mL | |
| 10 mM | 0.2850 mL | 1.4250 mL | 2.8501 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04681105
Conditions:Recurrent Acute Leukemia|Recurrent B Acute Lymphoblastic Leukemia|Recurrent Blastic Plasmacytoid Dendritic Cell Neoplasm|Recurrent Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Recurrent Hairy Cell Leukemia|Recurrent Hematologic Malignancy|Recurrent Hodgkin Lymphoma|Recurrent T Acute Lymphoblastic Leukemia|Refractory Acute Leukemia|Refractory B Acute Lymphoblastic Leukemia|Refractory Blastic Plasmacytoid Dendritic Cell Neoplasm|Refractory Chronic Myelogenous Leukemia, BCR-ABL1 Positive|Refractory Hairy Cell Leukemia|Refractory Hematologic Malignancy|Refractory Hodgkin Lymphoma|Refractory T Acute Lymphoblastic Leukemia|Systemic MastocytosisLink: https://clinicaltrials.gov/ct2/show/NCT03368664
Conditions:Multiple SclerosisLink: https://clinicaltrials.gov/ct2/show/NCT04862585
Conditions:Anatomic Stage 0 Breast Cancer AJCC v8|Anatomic Stage I Breast Cancer AJCC v8|Anatomic Stage IA Breast Cancer AJCC v8|Anatomic Stage IB Breast Cancer AJCC v8|Anatomic Stage II Breast Cancer AJCC v8|Anatomic Stage IIA Breast Cancer AJCC v8|Anatomic Stage IIB Breast Cancer AJCC v8|Anatomic Stage III Breast Cancer AJCC v8|Anatomic Stage IIIA Breast Cancer AJCC v8|Anatomic Stage IIIB Breast Cancer AJCC v8|Anatomic Stage IIIC Breast Cancer AJCC v8|Anatomic Stage IV Breast Cancer AJCC v8|Breast Carcinoma|Prognostic Stage 0 Breast Cancer AJCC v8|Prognostic Stage I Breast Cancer AJCC v8|Prognostic Stage IA Breast Cancer AJCC v8|Prognostic Stage IB Breast Cancer AJCC v8|Prognostic Stage II Breast Cancer AJCC v8|Prognostic Stage IIA Breast Cancer AJCC v8|Prognostic Stage IIB Breast Cancer AJCC v8|Prognostic Stage III Breast Cancer AJCC v8|Prognostic Stage IIIA Breast Cancer AJCC v8|Prognostic Stage IIIB Breast Cancer AJCC v8|Prognostic Stage IIIC Breast Cancer AJCC v8|Prognostic Stage IV Breast Cancer AJCC v8
Title:Study of H2 Antagonist and a Proton Pump Inhibitor of Selpercatinib in Healthy Participants
Status:Completed
updateDate:2024-10-30
Ctid:NCT05338502
Link: https://clinicaltrials.gov/ct2/show/NCT05338502
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT03145012
Conditions:CancerLink: https://clinicaltrials.gov/ct2/show/NCT00838526
Conditions:Gastroesophageal Reflux Disease (GERD)Link: https://clinicaltrials.gov/ct2/show/NCT05298683
Conditions:Multiple Myeloma|Renal Impairment|Neoplasms, Plasma Cell|Neoplasms by Histologic Type|Neoplasms|Paraproteinemias|Blood Protein Disorders|Hematologic DiseasesLink: https://clinicaltrials.gov/ct2/show/NCT02999633
Conditions:T-cell Type Acute Leukemia-Precursor|T-lymphoblastic Lymphoma/LeukaemiaLink: https://clinicaltrials.gov/ct2/show/NCT04397445
Conditions:Ranitidine Adverse Reaction|Pharmacokinetics|Food-drug InteractionLink: https://clinicaltrials.gov/ct2/show/NCT00668317
Conditions:CoughLink: https://clinicaltrials.gov/ct2/show/NCT01332630
Conditions:Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT03468777
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT02441673
Conditions:Post Anaesthetic ShiveringLink: https://clinicaltrials.gov/ct2/show/NCT01896557
Conditions:Coronary Artery Disease|Drug Interaction PotentiationLink: https://clinicaltrials.gov/ct2/show/NCT03140033
Conditions:Hemorrhage PostpartumLink: https://clinicaltrials.gov/ct2/show/NCT01539655
Conditions:Medullary Thyroid CancerLink: https://clinicaltrials.gov/ct2/show/NCT03011463
Conditions:Pharmacokinetics|Inhibition Enzyme|Drug Interaction PotentiationLink: https://clinicaltrials.gov/ct2/show/NCT02441894
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT01392755
Conditions:Healthy VolunteerLink: https://clinicaltrials.gov/ct2/show/NCT02123498
Conditions:Eustachian Tube Dysfunction|Laryngopharyngeal RefluxLink: https://clinicaltrials.gov/ct2/show/NCT02205489
Conditions:Relapsing-remitting Multiple SclerosisLink: https://clinicaltrials.gov/ct2/show/NCT02733640
Conditions:Antiplatelet EffectLink: https://clinicaltrials.gov/ct2/show/NCT00839306
Conditions:Gastroesophageal Reflux Disease (GERD)Link: https://clinicaltrials.gov/ct2/show/NCT02555852
Conditions:Gastroesophageal Reflux Disease (GERD)|Community-acquired PneumoniaLink: https://clinicaltrials.gov/ct2/show/NCT01737840
Conditions:DyspepsiaLink: https://clinicaltrials.gov/ct2/show/NCT00247130
Conditions:Peptic UlcersLink: https://clinicaltrials.gov/ct2/show/NCT02197143
Conditions:DyspepsiaLink: https://clinicaltrials.gov/ct2/show/NCT02172417
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT02170792
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT00131248
Conditions:Gastroesophageal RefluxLink: https://clinicaltrials.gov/ct2/show/NCT01908257
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT00527878
Conditions:JOB's Syndrome|Hyper-IgE Recurrent Infection Syndrome|Immune DeficiencyLink: https://clinicaltrials.gov/ct2/show/NCT00585351
Conditions:StrokeLink: https://clinicaltrials.gov/ct2/show/NCT01682408
Conditions:PharmacokineticsLink: https://clinicaltrials.gov/ct2/show/NCT00030992
Conditions:Renal Cell CarcinomaLink: https://clinicaltrials.gov/ct2/show/NCT01538797
Conditions:Reflux OesophagitisLink: https://clinicaltrials.gov/ct2/show/NCT00633672
Conditions:NSAID Associated Gastric UlcersLink: https://clinicaltrials.gov/ct2/show/NCT00401752
Conditions:Gastric UlcerLink: https://clinicaltrials.gov/ct2/show/NCT01131702
Conditions:HealthyLink: https://clinicaltrials.gov/ct2/show/NCT00633412
Conditions:NSAID Associated Gastric UlcersLink: https://clinicaltrials.gov/ct2/show/NCT00702871
Conditions:Ventilator Associated Pneumonia|Etiological Organisms|Antimicrobial Drug Susceptibility Pattern|Stress Ulcer ProphylaxisLink: https://clinicaltrials.gov/ct2/show/NCT00590928
Conditions:Critically Ill Patients|Indication for Stress Ulcer ProphylaxisLink: https://clinicaltrials.gov/ct2/show/NCT00247715
Conditions:Dyspepsia|Gastrointestinal DiseasesLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-001291-12
Condition:During cardiac surgery involving the use of cardiopulmonary bypass histamine is released in the blood. Histamine release has been related to an increased incidence of perioperative dysrhythmias and may be involved in other complications.Our aim is to find out whether the use of histamine blocking drugs will prevent these arrhythmias.The study population will consist of adult patients who must undergo elective cardiac surgery in which the use of cardiopulmonary bypass is necessaryInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
