| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
JAK
|
|---|---|
| 体外研究 (In Vitro) |
托法替尼前药-1(化合物 20g)(1 mM;37 °C 12 小时)在模拟胃液和肠液中从 0 到 12 小时没有明显降解[1]。
|
| 体内研究 (In Vivo) |
托法替布前药-1(22.5 mg/kg;口服)可通过将母体药物托法替布缓慢释放到循环中来减少托法替布的全身暴露。托法替尼前药-1(22.5 mg/kg;口服)可增加肠道暴露量,从而提高托法替尼的治疗效果。 Tofacitinib Prodrug-1(1.5 mg/kg;口服;每天两次,持续 4 天)可以有效减轻恶唑酮诱导的小鼠结肠炎。托法替尼前药-1(1.5 mg/kg;ig;每天两次,持续 4 天)对正常小鼠的全身免疫抑制没有明显影响,这可以降低与托法替尼相关的感染风险。托法替尼前药-1(2000 mg/kg;ig;单次)毒性低,口服剂量2000 mg/kg即可耐受,生化参数和器官指标未见明显变化。
|
| 酶活实验 |
前药在模拟胃液和肠液中的稳定性[1]
通过将胃蛋白酶加入pH=1.2的HCl水溶液中并将胰蛋白酶加入pH=6.8的磷酸盐缓冲液中来制备模拟胃液和肠液。化合物9和20a-20g的10mM储备溶液在DMSO中制备。通过将10μL储备溶液加入模拟胃液(990μL)或模拟肠液(990μL)中开始反应。然后,将混合物在37°C下孵育。最后,通过加入1mL含有内标(IS)柳氮磺胺吡啶(100μM)的冷却乙腈溶液,在0、2、5、8和12小时终止反应。将样品涡旋混合,在25°C下以10000 g离心10分钟,得到上清液,直接用于LC-MS/MS定量测定。流动相与水溶液实验部分前药的化学稳定性一致。梯度程序设置如下:0-10分钟5%A,5-10分钟95%A,10-15分钟5%A。流速为0.5 mL/min,根据标准曲线估算化合物的量。数据以平均值±SEM表示。 体外释放研究[1] 使用雄性Sprague-Dawley(SD)大鼠。通过将雄性SD大鼠的新鲜结肠内容物悬浮在含有0.1 M PBS和1.5 mg/mL d-葡萄糖(pH 6.8,之前用氮气鼓泡)的缓冲溶液中,制备结肠内容物的悬浮液,得到10重量%的悬浮液。在DMSO中制备化合物9和20a-20g的10mM储备溶液以及阳性对照柳氮磺胺吡啶。通过将每个样品的储备溶液(10μL)加入0.1 M PBS缓冲液(990μL)和10 wt%结肠内容物悬浮液(1 mL)中开始反应。然后,在氮气气氛中在37°C下孵育样品。最后,通过加入6mL IS(柳氮磺胺吡啶,100nM,溶于乙腈)的冷却溶液,在0、0.5、1、1.5、2、3、4、5、6、7和8小时终止反应。将样品涡旋混合并离心(8000 g,10分钟,4°C),冷冻储存(<-15°C)直至分析,以获得上清液,直接用于HPLC定量测定。梯度程序设置与前药在水溶液中的化学稳定性实验部分一致。流速为1.0 mL/min。柱温保持在25°C,根据标准曲线估算化合物的量。通过GraphPad软件计算体外释放AUC(托法替尼,0-5h)值和释放速率(托法替尼,0-2h)值。数据以平均值±SEM表示。 |
| 动物实验 |
PK Studies[1]
Male SD rats (aged 5–6 weeks, body weight range of 220–250 g, n = 6) were used, and they had free access to food and water and were maintained on a 12-h light/dark cycle in a temperature and humidity-controlled room for 3 days. After fasting for 12 h with free access to water, the rats were randomly divided into two groups. Tofacitinib and compound 20g were suspended in a 0.5% CMC-Na solution, and the rats in both groups were orally administered by gavage with tofacitinib (15 mg/kg) or compound 20g (22.5 mg/kg, equivalent to the dose of tofacitinib), respectively. Food was returned to rats 1 h after the oral administration. Blood samples of the tofacitinib group were collected into heparinized tubes via orbital vein bleeding at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 7 h, 10 h, and 12 h postdosing, and blood samples of the compound 20g group were gathered at 1, 2, 4, 6, 8, 12, 14, and 20 h postdosing. Plasma was separated by centrifugation (8000 g, 10 min, 4 °C) and stored frozen (<−15 °C) until analysis. The resulting plasma samples (50 μL) were extracted by adding a cooled solution of 150 μL of acetonitrile containing 10 μL of the IS, and these mixtures were further vortex-mixed and centrifuged at 8000g at 4 °C for 10 min. The resulting supernatants were directly transferred to the autosampler vial for LC–MS/MS analysis. Data were presented as means ± SEM. Intestinal Tissue Distribution Study[1] Male BALB/c mice (aged 5–6 weeks, body weight range: 25–30 g, n = 6) were were kept in a temperature- and humidity-controlled room with a 12-h light/dark cycle and provided free access to food and water for 3 days. After fasting for 12 h with free access to water, the mice were randomly divided into the tofacitinib and compound 20g groups. The tested compounds were dissolved in the 0.5% CMC-Na solution to prepare the suspensions. Then, the mice were orally administered by gavage with tofacitinib (15 mg/kg) or compound 20g (22.5 mg/kg), and food was returned to mice 1 h after the oral administration. The blood samples were collected via orbital vein bleeding at 0.5, 1, 2, 3, 4, 6, 9, and 12 h postdosing, and the intestinal tissues (duodenum, jejunum, ileum, and colon) were collected from the mice after sacrificing them at 0.5, 1, 2, 3, 4, 6, 9, and 12 h postdosing. The collected blood samples were added into heparinized tubes and further centrifuged (8000g, 10 min, 4 °C) to prepare plasma. The collected intestinal tissues from the mice were cut into pieces with scissors. The resultant pieces were weighed, diluted with six times the corresponding blank tissue pieces (weight/weight: w/w), and homogenated using a Benchmark D1000 rotor-stator hand homogenizer (Benchmark Scientific, New Jersey, US) at a speed of 10,000 rpm (2 × 30 s). Then, 50 μL of plasma and tissue homogenate samples was extracted with 150 μL of acetonitrile containing 10 μL of the IS, followed by vortexing and centrifugation (8000 g, 10 min, 4 °C). The resulting supernatants were directly used for LC–MS/MS analysis. Data were presented as means ± SEM. Oxazolone-Induced Colitis Model[1] Male BALB/C mice (aged 5–6 weeks, body weight range: 25–28 g, n = 7–9) had free access to food and water, and they were maintained in a 12 h light/dark cycle in a temperature- and humidity-controlled room for 3 days before the experiment. The mice were randomly divided into the model group, blank group, tofacitinib group (10 mg/kg), and compound 20g group (1.5 mg/kg). Among them, the oral dose of compound 20g (1.5 mg/kg) was equivalent to one-tenth dose of tofacitinib (10 mg/kg). On day 0, an approximately 2 cm × 2 cm field was shaved on the skin of the back of the mice with an electric clipper. Caution was taken to avoid open wounds. Then, the oxazolone, tofacitinib, and 20g groups were presensitized with oxazolone (200 μL, 3%, 4:1 ethanol/olive) by skin painting. Then, on the 5th day, the mice in each group were orally administered twice daily with the 0.5% CMC-Na solution, tofacitinib (10 mg/kg), or compound 20g (1.5 mg/kg) for 4 consecutive days. Subsequently, the mice were intrarectally administered with oxazolone (100 μL, 0.8%) to induce the colitis model on the seventh day. On the eighth day, the body weights of all mice (containing blank group) were examined and recorded twice a day after inducing the colitis model. Moreover, the stool consistency and presence of hematochezia were examined and recorded. All mice were sacrificed on the ninth day, and the colons were collected to examine their lengths and weights. The colons were prepared in a “Swiss roll” configuration, fixed in 4% PFA for 48 h, and further embedded in paraffin. The 5 μm cross-sections were used for histological evaluation. In addition, the spleens were gathered to examine spleen weight and calculate the spleen factor. The DAI was calculated as the sum of three subscores: loss of body weight (0 = none, 1 = 1–5%, 2 ≥5–10%, 3 ≥10–20%, 4 ≥20%), diarrheal score (0 = normal, 2 = loose, 4 = diarrhea), and bloody stool score (0 = normal, 2 = fecal occult blood, 4 = gross bleeding). The colon density was calculated as the colon weight/colon length. The histological scores were determined according to the histological grading criteria (inflammation: 0 none, 1 slight, 2 moderate; edema: 0 none, 1 slight, 2 moderate). The spleen index was calculated according to the following formula: spleen index (mg/g) = spleen weight (mg)/animal body weight (g). GraphPad Prism software was used for the statistical analysis of different groups. Data were presented as means ± SEM. Student’s t-test was used to compare the model and blank groups, and ANOVA followed by Fisher’s least significant difference (LSD) posthoc test was used to compare the model, tofacitinib, and compound 20g groups. Statistical significance between groups was considered at P < 0.05. Systemic Immunosuppression Study[1] Male BALB/c mice were used (aged 5–6 weeks, body weight range: 25–28 g, n = 10). The mice had free access to food and water and were maintained on a 12 h light/dark cycle in a temperature- and humidity-controlled room for 3 days before the experiment. Then, the mice were randomly divided into three groups, namely, tofacitinib, compound 20g, and blank groups. The same oral doses (tofacitinib: 10 mg/kg; compound 20g: 1.5 mg/kg) as the oxazolone-induced colitis model were selected for intragastric administration of mice twice daily for 4 days. Subsequently, the mice were sacrificed via neck dislocation 1 h after the last oral administration. The weights of the body and spleens were examined to calculate the spleen index [spleen index (mg/g) = spleen weight (mg)/animal body weight (g)], and spleens were crushed and diluted with 10 mL of PBS immediately. The resulting spleen cell samples (100 μL) were stained by adding 2 μL of the fluorophore-labeled antibody (APC-CD49b; BioLegend Biosciences) and 2 μL of the fluorophore-labeled antibody (PE-CD3; BioLegend Biosciences). Then, they were incubated at 4 °C for 1 h and diluted with 900 μL of PBS. The lymphocytes were first distinguished from cell debris on the basis of forward and side scatter properties (FSC-A and SSC-A), and CD49+ NK cells were then gated on the CD49+/CD3– quadrant The percentage of CD49+ NK cells in the staining samples was determined by flow cytometry (Becton Dickinson Calibur). The number of spleen cells in a 30 s time period was determined by flow cytometry with a flow rate of 35 μL/min (absolute spleen cell = 10 mL × 2/35 μL × number of spleen cells in 30 s, and absolute number of NK cell = absolute spleen cell × percentage of CD49+ NK cells). Acute Oral Toxicity Study[1] Healthy Kunming mice of both sexes (18–22 g; n = 10) were used. The mice had free access to food and water and were maintained on a 12 h light/dark cycle in a temperature- and humidity-controlled room for 1 week. After fasting for 12 h with free access to water prior to the experiment, four groups of animals (male control group, female control group, male test group, and female test group; n = 8) were used for the acute oral toxicity study. The control groups were treated with the 0.5% CMC-Na, and the test groups were treated with a single dose (2000 mg/kg) of prodrug 20g, which was suspended in the 0.5% CMC-Na solution. All treatments were intragastrically administered immediately after 12 h of fasting. The mice were observed continuously for any signs and symptoms of toxicity, and the body weight of these mice was monitored every day over the 7 day period after treatment. On the seventh day, whole blood samples of all mice were collected via orbital vein bleeding, and the collected blood samples were centrifuged at 3000g at 4 °C for 10 min. The ALT, AST, urea, and creatine levels in serum were determined with the assay kits, in accordance with the manufacturer’s instructions. All mice were sacrificed, and blood vessel catheter bleeding was performed with PBS. Subsequently, the liver, kidney, and spleen were collected and weighed, and the organ index was calculated as follows: organ index = weight of the experimental organ (mg)/weight of the experimental animal (g). |
| 参考文献 | |
| 其他信息 |
To mitigate the systemic adverse effects of tofacitinib, we constructed 5-ASA-PABA-MAC and 5-ASA-PABA-diamine colon-targeted delivery systems and synthesized tofacitinib azo prodrugs 9 and 20a-20g based on these systems. Release studies demonstrated that these systems efficiently release tofacitinib in vitro, with the 5-ASA-PABA-diamine system successfully achieving colon-targeted delivery of tofacitinib in vivo. Specifically, compared to an equimolar oral dose of tofacitinib, compound 20g showed a 3.67-fold reduction in plasma AUC (tofacitinib, 0-∞) and a 9.61-fold increase in colonic AUC (tofacitinib, 0-12h). Furthermore, mouse models showed that compound 20g (1.5 mg/kg) was substantially as effective as tofacitinib (10 mg/kg) in treating ulcerative colitis without impairing natural killer cells. These results suggest that compound 20g can serve as an effective alternative to tofacitinib, reducing its systemic adverse effects. Meanwhile, the 5-ASA-PABA-MAC and 5-ASA-PABA-diamine systems have also been shown to be effective for colon-targeted drug delivery. [1]
|
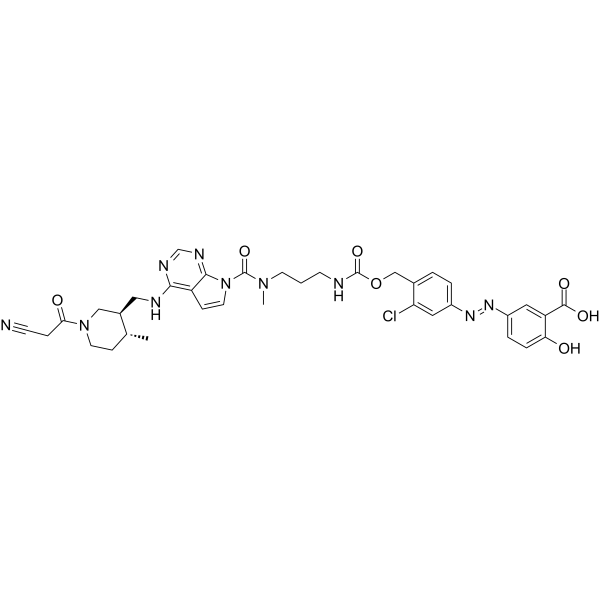
| 分子式 |
C36H39CLN10O7
|
|---|---|
| 分子量 |
759.21
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.3172 mL | 6.5858 mL | 13.1716 mL | |
| 5 mM | 0.2634 mL | 1.3172 mL | 2.6343 mL | |
| 10 mM | 0.1317 mL | 0.6586 mL | 1.3172 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
