| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| Other Sizes |
|
| 靶点 |
HMG-CoA reductase; HMG-CoA/3-hydroxy-3-methylglutaryl coenzyme A
|
|---|---|
| 体外研究 (In Vitro) |
阿托伐他汀治疗通过下调心肌梗死期间心肌细胞中 GRP78、caspase-12 和 CHOP 的表达,降低心肌细胞凋亡。此外,它还刺激内质网 (ER) 以应对心力衰竭和血管紧张素 II (Ang II) 刺激。 ) 紧张 [4]。
|
| 体内研究 (In Vivo) |
用电子压力计评价口服阿托伐他汀对小鼠脚爪炎性机械性高痛觉的影响。ELISA和RIA检测细胞因子和PGE(2)。 关键结果:在致敏动物中,阿托伐他汀治疗3天,剂量依赖性地减少了脂多糖(LPS)或抗原激发后引起的高痛觉。阿托伐他汀预处理可降低缓激肽和细胞因子(tnf - α、il -1 β和KC)诱导的高痛觉,以及脂多糖诱导的足跖皮肤il -1 β和PGE(2)的释放。在不影响血清胆固醇水平的情况下,甲羟戊酸联合治疗可阻止阿托伐他汀对lps诱导的高痛觉的抗痛觉作用。阿托伐他汀可抑制PGE诱导的高痛觉(2),提示阿托伐他汀的细胞内抗痛觉机制。阿托伐他汀对LPS或PGE(2)诱导的高痛觉的抗痛觉作用可以通过一氧化氮合酶(NOS)的非选择性抑制剂来阻止,但不能通过选择性抑制可诱导的NOS或缺乏这种酶的小鼠来阻止。[1]
|
| 细胞实验 |
细胞增殖测定基本上如前所述进行。简言之,将来自5名不同患者的SV-SMC以全生长培养基中每孔1×104个细胞的密度接种到24孔细胞培养板中。将细胞孵育过夜,然后在无血清培养基中静置3天,然后转移到含有5种不同浓度他汀类药物的全生长培养基(10%FCS)中。所有他汀类药物都在每个患者的细胞上进行了测试。2天后更换培养基和药物,4天后使用台盼蓝和血细胞仪在一式三个孔中测定活细胞数。细胞数的增加是通过从最终细胞数(第4天)中减去起始细胞数(0天)来计算的。然后将数据标准化为对照值(无他汀类药物),以校正来自不同患者的细胞之间增殖率的差异[2]。
|
| 动物实验 |
阿托伐他汀对脂多糖(LPS)或抗原刺激诱导的痛觉过敏的影响[1] 为了研究阿托伐他汀对脂多糖(LPS)诱导的炎症性痛觉过敏的影响,小鼠预先口服给予阿托伐他汀(剂量分别为1、3、10、30和90 mg kg⁻¹)或载体(PBS),每日一次,连续3天。在最后一次给予阿托伐他汀2小时后,小鼠接受足底注射LPS(100 ng paw⁻¹)或生理盐水(LPS的载体)。此外,部分小鼠在LPS刺激前1或2天接受阿托伐他汀(30 mg kg⁻¹)治疗。分别在LPS或生理盐水足底注射后0.5、1、3、5、7和24小时评估痛觉过敏反应。此外,我们还研究了阿托伐他汀对经mBSA致敏并接受抗原刺激的小鼠免疫炎症性痛觉过敏的影响。小鼠连续3天每日一次口服阿托伐他汀(30 mg kg⁻¹)或PBS进行预处理。末次给药2小时后,小鼠接受足底注射mBSA(90 μg paw⁻¹)或生理盐水。对照组小鼠(见上文)的足底注射mBSA。小鼠禁食8小时后接受阿托伐他汀或PBS。在抗原刺激后1、3和5小时评估痛觉过敏反应。
|
| 药代性质 (ADME/PK) |
阿托伐他汀的药代动力学特征呈剂量依赖性和非线性。口服后吸收迅速。服用40 mg后,1-2小时即可达到血浆峰浓度28 ng/ml,AUC约为200 ng∙h/ml。阿托伐他汀在肠壁和肝脏经历广泛的首过代谢,导致其绝对口服生物利用度仅为14%。与早晨服药相比,晚上服药后血浆阿托伐他汀浓度较低(Cmax和AUC降低约30%)。然而,无论何时服药,LDL-C的降低幅度均相同。与食物同服会导致Tmax延长,Cmax和AUC降低。乳腺癌耐药蛋白(BCRP)是一种膜结合蛋白,在阿托伐他汀的吸收中起着重要作用。药理遗传学研究表明,BCRP基因c.421C>A单核苷酸多态性(SNP)与BCRP基因421AA基因型相关,携带421AA基因型的个体功能活性降低,且阿托伐他汀的AUC值比携带421CC基因型的对照个体高1.72倍。这对于药物疗效和毒性反应的个体差异具有重要意义,尤其值得注意的是,BCRP c.421C>A多态性在亚洲人群中的发生率高于白种人。其他受此多态性影响的他汀类药物包括氟伐他汀、辛伐他汀和瑞舒伐他汀。由SCLCO1B1基因(溶质载体有机阴离子转运蛋白家族成员1B1)编码的肝脏转运蛋白OATP1B1(有机阴离子转运多肽1B1)的遗传差异已被证实会影响阿托伐他汀的药代动力学。对编码OATP1B1的基因(SLCO1B1)中c.521T>C单核苷酸多态性(SNP)的药理遗传学研究表明,与521TT纯合子个体相比,521CC纯合子个体的阿托伐他汀AUC增加了2.45倍。其他受此多态性影响的他汀类药物包括辛伐他汀、匹伐他汀、瑞舒伐他汀和普伐他汀。
消除途径 阿托伐他汀及其代谢物主要经胆汁排泄,不发生肠肝循环。阿托伐他汀的肾脏清除率极低,仅占清除剂量的不到1%。 分布容积 据报道,阿托伐他汀的分布容积为380升。 清除率 已记录的阿托伐他汀总血浆清除率为625毫升/分钟。 /乳汁/ 在另一项实验中,分别于妊娠第19天或哺乳第13天向雌性Wistar大鼠单次给予10毫克/公斤阿托伐他汀,结果表明该药物可通过胎盘转运并排泄到乳汁中。PMID:9520344 立普妥及其代谢物主要经肝脏和/或肝外代谢后通过胆汁排泄;然而,该药物似乎不会发生肠肝循环。口服立普妥后,尿液中回收的剂量不足2%。 /乳汁/ 尚不清楚阿托伐他汀是否会分泌到人乳中,但同类药物中有少量会进入母乳。哺乳期幼鼠血浆和肝脏中的药物浓度分别为其母乳中药物浓度的50%和40%。 立普妥的平均分布容积约为381升。立普妥与血浆蛋白的结合率≥98%。血液/血浆比值约为 0.25 表明药物穿透红细胞的能力较差。 有关阿托伐他汀(共 8 项)的更多吸收、分布和排泄(完整)数据,请访问 HSDB 记录页面。 查看更多代谢/代谢物 生物半衰期 阿托伐他汀的半衰期为14小时,而其代谢物的半衰期可达30小时。 /乳汁/……给哺乳期大鼠服用后,乳汁中的放射性在6.0小时达到最大值17.1 ng当量/mL,之后以7.8小时的半衰期下降。Nemoto H等; 《药理与医疗》26(7): 79-96 (1998) 立普妥在人体内的平均血浆消除半衰期约为14小时,但由于活性代谢物的贡献,其对HMG-CoA还原酶的抑制活性半衰期为20至30小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
毒性概述
识别和用途:阿托伐他汀是一种降胆固醇药物和羟甲基戊二酰辅酶A还原酶抑制剂。人体暴露和毒性:服用他汀类药物(包括阿托伐他汀)的患者中,罕见有致命性和非致命性肝功能衰竭的病例报告。服用他汀类药物(包括阿托伐他汀)的患者中,也罕见有因肌红蛋白尿继发横纹肌溶解症和急性肾功能衰竭的病例报告。由于胆固醇及其衍生物是胎儿正常发育所必需的,因此降脂药物在妊娠期间并无益处。动脉粥样硬化是一个慢性过程,妊娠期间停用降脂药物对原发性高胆固醇血症治疗的长期疗效影响甚微。神经精神反应的发生与他汀类药物治疗相关。这些反应包括行为改变、认知和记忆障碍、睡眠障碍和性功能障碍。动物研究:在一项为期两年的大鼠致癌性研究中,分别以10、30和100 mg/kg/天的剂量给药,在高剂量组雌性大鼠的肌肉中发现了2例罕见肿瘤:一例为横纹肌肉瘤,另一例为纤维肉瘤。在以10、40或120 mg/kg剂量给药两年的犬中,阿托伐他汀未对精液参数或生殖器官组织病理学产生不良影响。在交配前11周,以100 mg/kg/天的剂量给药的雄性大鼠,其精子活力和精子头部浓度降低,畸形精子数量增加。在剂量高达175 mg/kg的大鼠中进行的研究表明,阿托伐他汀对生育能力没有影响。 10只大鼠连续3个月接受100 mg/kg/天的阿托伐他汀治疗,其中2只出现附睾发育不全和无精子症;30 mg/kg和100 mg/kg剂量组的睾丸重量显著降低,100 mg/kg剂量组的附睾重量也较低。一项研究中,从妊娠第7天到哺乳第21天(断奶),分别给予大鼠20、100或225 mg/kg/天的阿托伐他汀,结果显示,225 mg/kg/天剂量组母鼠所产幼鼠在出生、新生儿期、断奶期和成熟期的存活率均降低。100 mg/kg/天剂量组母鼠所产幼鼠在出生后第4天和第21天体重下降;225 mg/kg/天剂量组母鼠所产幼鼠在出生时以及出生后第4天、第21天和第91天体重均下降。幼鼠发育迟缓。体外试验表明,无论是否进行代谢活化,阿托伐他汀在以下试验中均未表现出致突变性或致染色体断裂性:沙门氏菌和大肠杆菌的Ames试验、中国仓鼠肺细胞的HGPRT正向突变试验以及中国仓鼠肺细胞的染色体畸变试验。体内小鼠微核试验结果也为阴性。 阿托伐他汀选择性地竞争性抑制肝脏酶HMG-CoA还原酶。由于HMG-CoA还原酶负责在胆固醇生物合成途径中将HMG-CoA转化为甲羟戊酸,因此抑制HMG-CoA还原酶可导致肝脏胆固醇水平下降。肝脏胆固醇水平降低会刺激肝脏低密度脂蛋白胆固醇(LDL-C)受体上调,从而增加肝脏对LDL-C的摄取并降低血清LDL-C浓度。 肝毒性 阿托伐他汀治疗与1%至3%的患者出现轻度、无症状且通常短暂的血清转氨酶升高相关,但不到1%的患者会出现ALT水平超过正常值上限(ULN)3倍的情况。在前瞻性监测的大规模研究的汇总分析中,接受阿托伐他汀治疗的患者中ALT水平超过正常值上限3倍的比例为0.7%,而安慰剂组为0.3%。这些升高在高剂量阿托伐他汀治疗中更为常见,每日80毫克剂量组的发生率为2.3%。大多数升高均为自限性,无需调整剂量。 阿托伐他汀也与明显的、临床上可观察到的肝损伤相关,但这种情况罕见,发生率约为 1/3000 至 1/5000 的治疗患者。阿托伐他汀肝毒性的临床表现差异很大,从单纯性胆汁淤积性肝炎到混合型,再到明显的肝细胞损伤。损伤发生的潜伏期也差异很大,从 1 个月到数年不等。然而,大多数病例发生在开始服用阿托伐他汀后 6 个月内或剂量增加后数月内。最常见的表现是胆汁淤积性肝炎,其严重程度通常为轻度至中度,且具有自限性(病例 1 和 2)。阿托伐他汀肝毒性也可表现为明显的肝细胞损伤模式,伴有血清转氨酶水平显著升高,而碱性磷酸酶水平几乎没有升高或没有升高。皮疹、发热和嗜酸性粒细胞增多症并不常见,但至少三分之一的肝细胞癌病例具有自身免疫特征,表现为免疫球蛋白水平升高、抗核抗体(ANA)阳性以及肝活检证实为自身免疫性肝炎(病例3和4)。这些自身免疫性病例通常在停用阿托伐他汀后缓解,但有时可能需要糖皮质激素治疗才能痊愈。然而,值得注意的是,一些由阿托伐他汀引起的疑似自身免疫性肝炎病例在停药后并未缓解,而是持续存在,需要长期免疫抑制治疗。目前尚不清楚这些持续性自身免疫性肝炎病例是由他汀类药物治疗引起的,还是由易感宿主服用他汀类药物诱发的。另一种可能性是,这种关联纯属巧合,代表服用他汀类药物的人新发自身免疫性肝炎。 可能性评分:A(临床上明显的肝损伤的已知原因)。 查看更多妊娠和哺乳期影响 蛋白结合 阿托伐他汀与血浆蛋白高度结合,超过98%的给药剂量以结合形式存在。 |
| 参考文献 |
[1]. Santodomingo-Garzón T, et al. Atorvastatin inhibits inflammatory hypernociception. Br J Pharmacol. 2006 Sep;149(1):14-22.
[2]. Turner NA, et al. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007 Oct;50(4):458-61. [3]. Nawrocki, J.W., et al., Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler Thromb Vasc Biol, 1995. 15(5): p. 678-82. [4]. Song XJ, et al. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci. 2011;8(7):564-72. [5]. Li Y, et al. Inhibition of endoplasmic reticulum stress signaling pathway: A new mechanism of statins to suppress the development of abdominal aortic aneurysm. PLoS One. 2017 Apr 3;12(4):e0174821. [6]. Ming-Bai Hu, et al. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct Pathol. Sep-Oct 2018;42(5):409-415. [7]. In Vitro Screening for β-Hydroxy-β-methylglutaryl-CoA Reductase Inhibitory and Antioxidant Activity of Sequentially Extracted Fractions of Ficus palmata Forsk. Biomed Res Int. 2014; 2014: 762620. |
| 其他信息 |
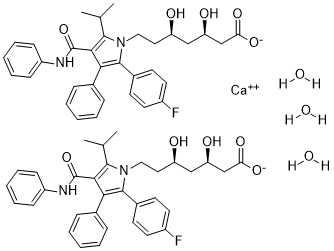
阿托伐他汀钙三水合物是阿托伐他汀钙的三水合物形式。它是一种环境污染物和外源性物质。它是一种水合物,也是一种合成的他汀类药物。它含有阿托伐他汀钙。
阿托伐他汀钙是阿托伐他汀的钙盐,阿托伐他汀是一种合成的降脂药。阿托伐他汀竞争性抑制肝脏羟甲基戊二酰辅酶A (HMG-CoA) 还原酶,该酶催化HMG-CoA转化为甲羟戊酸,这是胆固醇合成的关键步骤。该药物增加肝细胞表面低密度脂蛋白 (LDL) 受体的数量,增强LDL的摄取和分解代谢,减少LDL的生成和LDL颗粒的数量,并降低血浆胆固醇和脂蛋白水平。与其他他汀类药物一样,阿托伐他汀也可能具有直接的抗肿瘤活性,其机制可能是通过抑制小GTP结合蛋白等蛋白质的法尼基化和香叶基香叶基化,从而导致细胞周期停滞在G1期。该药物还可能通过mTOR依赖性抑制Akt磷酸化,增强肿瘤细胞对细胞抑制剂的敏感性。 阿托伐他汀是一种吡咯和庚酸衍生物,属于羟甲基戊二酰辅酶A还原酶抑制剂(他汀类药物),也是一种降胆固醇药物,用于降低血清中低密度脂蛋白胆固醇、载脂蛋白B和甘油三酯的水平。本品用于治疗高脂血症,提高血清高密度脂蛋白胆固醇(HDL-C)水平,并用于预防具有多种危险因素患者的心血管疾病。 另见:阿托伐他汀(含活性成分);阿托伐他汀钙三水合物;依折麦布(成分之一)。 药物适应症 单纯性高胆固醇血症(杂合子、纯合子或其他原发性高胆固醇血症)、混合型高脂血症;预防心血管事件 |
| 分子式 |
C66H74CAF2N4O13
|
|---|---|
| 分子量 |
1209.3876
|
| 精确质量 |
1208.484
|
| 元素分析 |
C, 65.55; H, 6.17; Ca, 3.31; F, 3.14; N, 4.63; O, 17.20
|
| CAS号 |
344423-98-9
|
| 相关CAS号 |
134523-03-8 (calcium);344423-98-9 (calcium trihydrate);134523-00-5 (free acid);134523-01-6 (sodium); 874114-41-7 (magnesium);
|
| PubChem CID |
656846
|
| 外观&性状 |
Typically exists as White to off-white solid at room temperature
|
| LogP |
9.91
|
| tPSA |
256.93
|
| 氢键供体(HBD)数目 |
9
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
22
|
| 重原子数目 |
86
|
| 分子复杂度/Complexity |
817
|
| 定义原子立体中心数目 |
4
|
| SMILES |
[Ca+2].FC1C([H])=C([H])C(=C([H])C=1[H])C1=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C(C(N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)=C(C([H])(C([H])([H])[H])C([H])([H])[H])N1C([H])([H])C([H])([H])[C@]([H])(C([H])([H])[C@]([H])(C([H])([H])C(=O)[O-])O[H])O[H].FC1C([H])=C([H])C(=C([H])C=1[H])C1=C(C2C([H])=C([H])C([H])=C([H])C=2[H])C(C(N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)=C(C([H])(C([H])([H])[H])C([H])([H])[H])N1C([H])([H])C([H])([H])[C@]([H])(C([H])([H])[C@]([H])(C([H])([H])C(=O)[O-])O[H])O[H].O([H])[H].O([H])[H].O([H])[H]
|
| InChi Key |
SHZPNDRIDUBNMH-NIJVSVLQSA-L
|
| InChi Code |
InChI=1S/2C33H35FN2O5.Ca.3H2O/c2*1-21(2)31-30(33(41)35-25-11-7-4-8-12-25)29(22-9-5-3-6-10-22)32(23-13-15-24(34)16-14-23)36(31)18-17-26(37)19-27(38)20-28(39)40/h2*3-16,21,26-27,37-38H,17-20H2,1-2H3,(H,35,41)(H,39,40)3*1H2/q+2/p-2/t2*26-,27-/m11..../s1
|
| 化学名 |
calcium
(3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-(phenylcarbamoyl)-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate
trihydrate
|
| 别名 |
liptonorm; CI-981; CI 981; CI981; Atorvastatin; atorvastatin calcium trihydrate; atorvastatin calcium trihydrate; 344423-98-9; Atorvastatin hemicalcium trihydrate; Totalip; Atorvastatin calcium salt trihydrate; ATORVASTATIN CALCIUM; Torvast; Atorvastatin calcium [USAN]; atorvastatin calcium salt
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.8269 mL | 4.1343 mL | 8.2686 mL | |
| 5 mM | 0.1654 mL | 0.8269 mL | 1.6537 mL | |
| 10 mM | 0.0827 mL | 0.4134 mL | 0.8269 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 奈莫雷生盐酸盐
奈莫雷生盐酸盐
 E55888 HCL
E55888 HCL
 FITC-Dextran (MW 110000)
FITC-Dextran (MW 2000000)
FITC-Dextran (MW 110000)
FITC-Dextran (MW 2000000)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
