| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Voltage-Gated Sodium Channels (NaV)
|
|---|---|
| 体外研究 (In Vitro) |
甲哌卡因通过与神经元细胞膜上特定的电压门控钠离子通道结合,抑制钠离子内流和膜去极化。这会阻碍神经冲动的起始和传导,从而导致暂时性感觉丧失。与其他局部麻醉药相比,甲哌卡因起效更快,作用时间适中[2]。甲哌卡因的作用时间中等,比普鲁卡因短,但起效较快(比普鲁卡因快)[3]。S(-)-布比卡因对TTXs Na(+)通道具有选择性,而甲哌卡因则表现出对Na(v)1.8通道的优先使用依赖性阻滞作用[4]。
甲哌卡因与神经元细胞膜上特定的电压门控钠离子通道结合,抑制钠离子内流和膜去极化。这导致神经冲动的产生和传导受阻,从而引起可逆性感觉丧失。 |
| 体内研究 (In Vivo) |
甲哌卡因在临床上用于局部麻醉,可通过浸润麻醉、神经阻滞和硬膜外麻醉实现。它起效迅速,作用持续时间中等(90-180分钟)。其化学结构与布比卡因相关,但药理作用与利多卡因相关。它常用于牙科手术和小型外科手术。
|
| 酶活实验 |
对于受体结合实验,纯化的电压门控钠通道(例如,来自大鼠脑的通道)被重组到脂质体或膜中。放射性配体结合实验使用[3H]箭毒蛙毒素或[3H]石房蛤毒素进行。将甲哌卡因以不同浓度(0.1-1000 uM)与膜孵育。过滤或离心后测量特异性结合。可以确定IC50值。或者,也可以使用异源表达的NaV通道进行电生理学实验。
|
| 细胞实验 |
在电生理实验中,表达人NaV1.7通道的HEK293细胞培养于含10%胎牛血清(FBS)的DMEM培养基中。进行全细胞膜片钳记录。外液包含NaCl、KCl、CaCl2、MgCl2、HEPES和葡萄糖。内液包含CsF、CsCl和EGTA。细胞钳制于-120 mV,然后阶跃至-10 mV。施加浓度为0.1-1000 uM的甲哌卡因,并测量峰值钠电流的降低。
|
| 动物实验 |
体内麻醉活性评估采用动物模型,例如大鼠甩尾试验或豚鼠皮内风团试验。将0.5-2%的甲哌卡因皮下或皮内注射。通过对针刺或热刺激(例如热板)的反应来测量感觉阻滞的程度和持续时间。对于神经阻滞模型,暴露坐骨神经并注射局部麻醉剂溶液;评估运动和感觉功能。
|
| 药代性质 (ADME/PK) |
吸收、分布和排泄
局部吸收:局部麻醉药的全身吸收率取决于给药的总剂量和浓度、给药途径、给药部位的血管分布以及麻醉溶液中肾上腺素的存在。局部麻醉药代谢迅速;仅有少量(5%至10%)以原形经尿液排出。肝脏是主要的代谢场所,超过50%的给药剂量以代谢物的形式经胆汁排出。代谢/代谢物:局部麻醉药代谢迅速;仅有少量(5%至10%)以原形经尿液排出。肝脏是主要的代谢场所,超过50%的给药剂量以代谢物的形式经胆汁排出。局部麻醉药代谢迅速;仅有少量(5%至10%)以原形经尿液排出。肝脏是主要的代谢场所,超过50%的给药剂量以代谢物的形式经胆汁排出。清除途径:该药物代谢迅速;仅有少量(5%至10%)麻醉剂以原形经尿液排出。肝脏是主要的代谢场所,超过50%的给药剂量以代谢物的形式经胆汁排出。半衰期:甲哌卡因在成人体内的半衰期为1.9至3.2小时,在新生儿体内的半衰期为8.7至9小时。 局部注射后,甲哌卡因迅速吸收。起效时间为3-5分钟,持续时间为90-180分钟(取决于剂量和是否同时使用血管收缩剂)。它主要在肝脏中通过CYP1A2和CYP3A4代谢为无活性代谢物。成人半衰期为1.9-3.2小时。血浆蛋白结合率约为75-80%。主要经肾脏排泄。它能透过胎盘,但中枢神经系统穿透性低。 |
| 毒性/毒理 (Toxicokinetics/TK) |
毒性概述
局部麻醉药通过阻断神经冲动的产生和传导发挥作用。其机制可能包括提高神经的电兴奋阈值、减慢神经冲动的传播速度以及降低动作电位的上升速率。通常,麻醉的进展与受影响神经纤维的直径、髓鞘化程度和传导速度有关。临床上,神经功能丧失的顺序如下:痛觉、温度觉、触觉、本体感觉和骨骼肌张力。妊娠和哺乳期影响 ◉ 哺乳期用药概述 目前尚无关于哺乳期使用甲哌卡因的信息。鉴于其他局部麻醉药在母乳中的分泌量较低,哺乳期单次使用甲哌卡因不太可能对母乳喂养的婴儿产生不良影响。但是,尤其对于母乳喂养的新生儿或早产儿,可能更倾向于选择其他药物。据报道,分娩时使用甲哌卡因作为局部麻醉剂可能会影响部分婴儿的初始母乳喂养行为,但不会影响产后前5天的体重增长。尽管关于甲哌卡因的研究有限,但似乎在良好的母乳喂养支持下,无论是否联合使用芬太尼或其衍生物,硬膜外麻醉对母乳喂养的成功率几乎没有或完全没有不良影响。分娩镇痛可能会延迟泌乳的开始。需要更多研究来阐明分娩时使用甲哌卡因对母乳喂养结局的影响。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对哺乳和母乳的影响 一项比较在正常分娩过程中使用甲哌卡因、布比卡因和利多卡因进行硬膜外镇痛效果的研究发现,在产后最初5天,三组母乳喂养婴儿的体重变化没有差异。所有组的总体体重增长均在正常范围内。 在分娩后一小时内接受甲哌卡因阴部神经阻滞的6名婴儿中,有4名婴儿开始母乳喂养的时间较晚,且初始乳汁量少于10名在分娩过程中未接受麻醉的婴儿。这些差异的长期后果尚未见报道。一项针对孕晚期至产后12个月的妇女及其婴儿的全国性调查比较了在分娩过程中接受和未接受镇痛药的母亲的第二阶段哺乳期持续时间。药物类别包括:单独使用脊髓或硬膜外麻醉、脊髓或硬膜外麻醉联合其他药物,以及单独使用其他镇痛药。接受任何类型药物的女性,其泌乳期延迟至第二阶段(>72小时)的风险约为未接受分娩镇痛的女性的两倍。蛋白结合率:甲哌卡因与血浆蛋白的结合率约为75%。通常,药物血浆浓度越低,其血浆蛋白结合率越高。毒性数据:在恒河猴中,甲哌卡因的平均致痫剂量为18.8 mg/kg,平均动脉血浆浓度为24.4 μg/mL。LD50:23-35 mg/kg(静脉注射,小鼠)(A308);LD50:280 mg/kg(皮下注射,小鼠)(A308)。 甲哌卡因在治疗剂量下通常耐受性良好。常见副作用包括剂量依赖性的中枢神经系统反应(头晕、嗜睡、意识模糊、癫痫发作)和心血管反应(低血压、心动过缓、心脏骤停)。高剂量可引起高铁血红蛋白血症。过敏反应罕见。过量用药可能致命。其毒性与其他酰胺类局部麻醉药相似。 |
| 参考文献 | |
| 其他信息 |
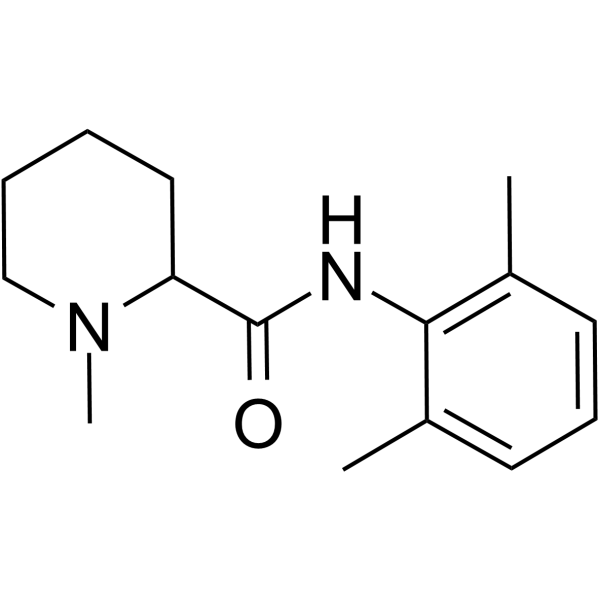
甲哌卡因是一种哌啶羧酰胺化合物,由N-甲基哌啶酸和2,6-二甲基苯胺通过酰胺键结合而成。它是一种局部酰胺类麻醉药。甲哌卡因既是局部麻醉药,也是一种药物过敏原。它的化学结构与布比卡因相关,但药理作用与利多卡因相关。它适用于浸润麻醉、神经阻滞和硬膜外麻醉。甲哌卡因仅在局部大剂量使用时有效,因此不应通过此途径使用。(摘自《美国医学会药物评价杂志》,1994年,第168页)甲哌卡因是一种酰胺类局部麻醉药。甲哌卡因的生理作用是通过局部麻醉实现的。甲哌卡因是一种酰胺类局部麻醉药。在注射部位,甲哌卡因与神经元细胞膜上特定的电压门控钠离子通道结合,抑制钠离子内流和膜去极化。这导致神经冲动的起始和传导受阻,从而引起可逆性感觉丧失。与其他局部麻醉药相比,该药起效更快,作用持续时间适中。甲哌卡因是一种局部麻醉药,其化学结构与布比卡因相关,但药理作用与利多卡因相关。它适用于浸润麻醉、神经阻滞和硬膜外麻醉。甲哌卡因仅在局部大剂量应用时有效,因此不应通过此途径使用。(摘自《美国医学会药物评价杂志》,1994年,第168页)另见:盐酸甲哌卡因(盐形式)。药物适应症:本品用于通过局部浸润、外周神经阻滞技术以及包括硬膜外阻滞和尾部阻滞在内的中枢神经系统技术,产生局部或区域镇痛和麻醉。FDA标签:作用机制:局部麻醉药通过阻断神经冲动的产生和传导发挥作用。其机制可能包括提高神经的电兴奋阈值、减缓神经冲动的传导速度以及降低动作电位的上升速率。通常,麻醉的进展与受累神经纤维的直径、髓鞘化程度和传导速度有关。临床上,神经功能丧失的顺序如下:痛觉、温度觉、触觉、本体感觉和骨骼肌张力。药效学:甲哌卡因是一种酰胺类局部麻醉药。甲哌卡因起效迅速,作用持续时间中等;其商品名为卡波卡因和泊洛卡因。甲哌卡因用于局部浸润和区域麻醉。局部麻醉药的全身吸收可影响心血管系统和中枢神经系统。正常治疗剂量下达到的血药浓度对心脏传导、兴奋性、不应期、收缩力和外周血管阻力的影响极小。
甲哌卡因(CAS:96-88-8)是一种经FDA批准的局部麻醉剂(商品名:卡波卡因、斯堪的纳)。它以消旋混合物的形式存在。与利多卡因不同,甲哌卡因不具有血管扩张作用,因此在不希望发生血管收缩的手术中,无需使用肾上腺素。它属于酰胺类麻醉剂,因此与酯类麻醉剂发生交叉过敏反应的情况很少见。参考文献:DrugBank DB00961。 |
| 分子式 |
C15H22N2O
|
|---|---|
| 分子量 |
246.35
|
| 精确质量 |
246.173
|
| CAS号 |
96-88-8
|
| 相关CAS号 |
(+)-Mepivacaine;24358-84-7;Mepivacaine hydrochloride;1722-62-9;Mepivacaine-d3;1346597-90-7
|
| PubChem CID |
4062
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.077 g/cm3
|
| 沸点 |
383.062ºC at 760 mmHg
|
| 熔点 |
150.5 °C
|
| 闪点 |
185.47ºC
|
| LogP |
2.737
|
| tPSA |
32.34
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
282
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC1=C(C(=CC=C1)C)NC(=O)C2CCCCN2C
|
| InChi Key |
INWLQCZOYSRPNW-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C15H22N2O/c1-11-7-6-8-12(2)14(11)16-15(18)13-9-4-5-10-17(13)3/h6-8,13H,4-5,9-10H2,1-3H3,(H,16,18)
|
| 化学名 |
N-(2,6-dimethylphenyl)-1-methylpiperidine-2-carboxamide
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 33.33 mg/mL (135.30 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0593 mL | 20.2963 mL | 40.5927 mL | |
| 5 mM | 0.8119 mL | 4.0593 mL | 8.1185 mL | |
| 10 mM | 0.4059 mL | 2.0296 mL | 4.0593 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04454203
Conditions:Tourniquet Hypertension|Intraoperative Hypertension|Total Ankle Arthroplasty|Ankle FusionLink: https://clinicaltrials.gov/ct2/show/NCT07239999
Conditions:Recovery Period|AnesthesiaLink: https://clinicaltrials.gov/ct2/show/NCT06005480
Conditions:Regional Anesthesia Morbidity|Pain, Acute|Healthy
Title:Genicular and Anterior Femoral Cutaneous Nerve Blocks for Total Knee Arthroplasty
Status:Completed
updateDate:2025-10-08
Ctid:NCT05980546
Link: https://clinicaltrials.gov/ct2/show/NCT05980546
Conditions:Total Knee Replacement|Genicular Nerve Block|Opioid UseLink: https://clinicaltrials.gov/ct2/show/NCT06291727
Conditions:Anesthesia, Spinal|Arthroplasty, Replacement, KneeLink: https://clinicaltrials.gov/ct2/show/NCT05425979
Conditions:Ankle Block|Foot SurgeryLink: https://clinicaltrials.gov/ct2/show/NCT05728151
Conditions:Pediatric Squint SurgeriesLink: https://clinicaltrials.gov/ct2/show/NCT06271174
Conditions:Persistent Postoperative PainLink: https://clinicaltrials.gov/ct2/show/NCT01860417
Conditions:Degenerative Disc Disease|Intervertebral Disc Disease|Low Back PainLink: https://clinicaltrials.gov/ct2/show/NCT05201313
Conditions:Vaginal Discharge|Perineum; Rupture|AnalgesiaLink: https://clinicaltrials.gov/ct2/show/NCT04257682
Conditions:Knee Osteoarthritis|Hip OsteoarthritisLink: https://clinicaltrials.gov/ct2/show/NCT03211949
Conditions:Nerve Block|Surgery|Anesthesia|Brachial Plexus BlockLink: https://clinicaltrials.gov/ct2/show/NCT03365752
Conditions:Knee ArthroscopyLink: https://clinicaltrials.gov/ct2/show/NCT05252117
Conditions:Contraception|IUD|Anesthesia, LocalLink: https://clinicaltrials.gov/ct2/show/NCT03838874
Conditions:Knee Osteoarthritis|Hip OsteoarthritisLink: https://clinicaltrials.gov/ct2/show/NCT04822415
Conditions:Symptomatic Irreversible PulpitisLink: https://clinicaltrials.gov/ct2/show/NCT04113954
Conditions:Compartment Syndrome of Leg|HealthyLink: https://clinicaltrials.gov/ct2/show/NCT03390426
Conditions:Tourniquet Hypertension|Intraoperative Hypertension|Total Ankle Arthroplasty|Ankle FusionLink: https://clinicaltrials.gov/ct2/show/NCT01574807
Conditions:PainLink: https://clinicaltrials.gov/ct2/show/NCT02406703
Conditions:Impact of Anesthetic Choice on CostsLink: https://clinicaltrials.gov/ct2/show/NCT02584452
Conditions:Rupture of Anterior Cruciate Ligament|Tear of Anterior Cruciate LigamentLink: https://clinicaltrials.gov/ct2/show/NCT01522534
Conditions:Injuries|Pain|EmergenciesLink: https://clinicaltrials.gov/ct2/show/NCT03942991
Conditions:Badly Decayed Lower First Primary Molars in ChildrenLink: https://clinicaltrials.gov/ct2/show/NCT02726620
Conditions:HypotensionLink: https://clinicaltrials.gov/ct2/show/NCT02078063
Conditions:Insertion of Intrauterine ContraceptionLink: https://clinicaltrials.gov/ct2/show/NCT00825786
Conditions:Local AnestheticLink: https://clinicaltrials.gov/ct2/show/NCT02980926
Conditions:Anesthesia, Spinal|Arthroplasty, Replacement, Knee|Pain Management|Early Ambulation|Ambulatory Surgical ProceduresLink: https://clinicaltrials.gov/ct2/show/NCT02384915
Conditions:Knee ArthritisLink: https://clinicaltrials.gov/ct2/show/NCT02433899
Conditions:Gingival RecessionLink: https://clinicaltrials.gov/ct2/show/NCT02110966
Conditions:Symptomatic Irreversible PulpitisLink: https://clinicaltrials.gov/ct2/show/NCT01981291
Conditions:Orthopedic Surgical Procedures|Postoperative PainLink: https://clinicaltrials.gov/ct2/show/NCT01485653
Conditions:Nerve Block|Neuromuscular Blockade|AnesthesiaLink: https://clinicaltrials.gov/ct2/show/NCT00802009
Conditions:AnesthesiaLink: https://clinicaltrials.gov/ct2/show/NCT01032798
Conditions:Local Anesthetic EffectivenessLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2016-002325-11
Condition:intra and post operative pain managementLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2015-001146-29
Condition:Degenerative spine pain when there are no indications for surgical treatmentDolor de patologia degenerativa de columna tratado de forma conservadoraLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2013-001795-38
Condition:Cranial facial painDolore cranio faccialeLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2012-004444-30
Condition:Treatment of degenerative disc diseaseTratamiento de la discopatía degenerativaLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2012-001704-38
Condition:Patients scheduled for single shot axillary brachial plexus block for hand, wrist, or forearm orthopedic surgeryLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-015867-15
Condition:Parkinson DeseaseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-004936-22
Condition:Regional anesthesia strategies during forefoot surgery. JNJ63955918
JNJ63955918
 Zocainone
Zocainone
 NVP-QBE170
NVP-QBE170
 B-GYKI-38233 hydrochloride
B-GYKI-38233 hydrochloride
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
