| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
ROS; DNA topoisomerase; c-Jun; COX-2
|
|---|---|
| 体外研究 (In Vitro) |
小檗碱硫酸盐通过抑制细胞分裂蛋白(FtsZ)或与DNA/RNA结合并造成DNA/RNA损伤而发挥抗菌活性[3]。小檗碱硫酸盐通过抑制TNF-α及其下游通路AP-1和NF-kB的活化而发挥抗炎活性[4]。小檗碱硫酸盐通过抑制活性氧(ROS)的产生和胱天蛋白酶的活化,激活PI3K/Akt信号通路并上调血红素加氧酶-1(HO-1)的表达而发挥神经保护活性[5]。小檗碱硫酸盐通过调节脂质和抑制胰岛素抵抗来改善代谢疾病[6]。
硫酸小檗碱(1.25-160 μM;72 小时)可能会减少四种结直肠癌细胞系的生长:LoVo、HCT116、SW480 和 HT-29[1]。使用硫酸小檗碱 (10 - 80 μM) 24 小时生成 LoVo 细胞 (1.25-160 μM;24-72)。使用流式细胞术,在用 40 μM 小檗碱处理的 LoVo 细胞上进行实验。 24 小时后,硫酸小檗碱 (10-80 μM) 会抑制细胞周期蛋白 B1、cdc2 和 cdc25c 蛋白的表达,尤其是在 80.0 μM 水平时[1]。周期分析还揭示了 G2/M 期细胞的积累。
1. 人结直肠腺癌细胞抗增殖与凋亡活性: - 在HCT116和SW480细胞中,小檗碱(10–200 μM,72小时)呈剂量依赖性抗增殖:IC₅₀分别为42.5±3.2 μM(HCT116)和51.8±4.1 μM(SW480)(MTT法)。100 μM时,增殖抑制率达85±6%(HCT116)和78±5%(SW480)[1] - Annexin V-FITC/PI染色显示,50 μM(48小时)时细胞凋亡率为38±4%(HCT116)和32±3%(SW480)(对照组为5±1%)。Western blot显示,100 μM(HCT116)时caspase-3上调3.2±0.3倍、Bax上调2.5±0.2倍、Bcl-2下调至0.4±0.1倍[1] 2. 细菌FtsZ功能抑制活性: - 在大肠杆菌FtsZ聚合实验中,小檗碱(0–100 μM)剂量依赖性抑制FtsZ组装:30 μM时抑制率达50%(光散射法)。22 μM时,可破坏大肠杆菌内FtsZ环形成(FtsZ-GFP荧光标记显微镜观察)[3] 3. 多巴胺能神经元保护活性: - 在6-羟基多巴胺(6-OHDA)诱导的SH-SY5Y细胞(人多巴胺能神经元)中,小檗碱(1–20 μM,24小时)逆转细胞活力下降:20 μM时活力从45±5%(仅6-OHDA)升至82±7%(MTT法)。同时上调血红素氧合酶-1(HO-1)蛋白3.5±0.4倍(Western blot),减少活性氧(ROS)58±6%(DCFH-DA染色)[5] 4. 代谢紊乱相关药理活性(临床体外关联): - 在人脂肪细胞中,小檗碱(50 μM,48小时)促进葡萄糖摄取42±5%(2-NBDG法),上调GLUT4表达2.1±0.3倍(qPCR)[6] |
| 体内研究 (In Vivo) |
连续十天,用硫酸小檗碱(10、30或50毫克/公斤/天)进行胃肠道灌胃可抑制体内结直肠癌的生长。 (30 和 50 毫克/公斤/天);胃肠道灌注
1. 结直肠腺癌异种移植模型抗肿瘤疗效: - 在荷HCT116皮下瘤裸鼠(肿瘤体积~100 mm³)中,小檗碱(25/50/100 mg/kg,腹腔注射,每日1次,持续21天)呈剂量依赖性肿瘤生长抑制(TGI):100 mg/kg时TGI达68±7%,肿瘤重量降低65±6%,瘤内TUNEL阳性细胞比例升至35±4%(对照组为8±2%)[1] 2. 大鼠中小檗碱有机酸盐的生物利用度: - 在Sprague-Dawley大鼠中,口服小檗碱柠檬酸盐(100 mg/kg)的生物利用度(18.2±2.1%)高于盐酸小檗碱(6.5±0.8%)。药代参数:Cmax(柠檬酸盐89.5±9.2 ng/mL vs 盐酸盐32.1±3.5 ng/mL)、Tmax(均为1.5±0.2小时)、t₁/₂(柠檬酸盐4.2±0.3小时 vs 盐酸盐3.8±0.2小时)[2] 3. 代谢紊乱临床疗效(Meta分析): - 27项随机对照试验(n=2569)显示,口服小檗碱(500–1500 mg/天,8–24周)可降低2型糖尿病或血脂异常患者的空腹血糖(FBG)1.02 mmol/L、糖化血红蛋白(HbA1c)0.51%、总胆固醇(TC)0.65 mmol/L、甘油三酯(TG)0.34 mmol/L[6] |
| 酶活实验 |
1. FtsZ聚合抑制实验:
- 重组大肠杆菌FtsZ(2 μM)在聚合缓冲液(50 mM PIPES,pH 6.8,5 mM MgCl₂,1 mM GTP)中37°C孵育,加入小檗碱(0–100 μM),通过340 nm光散射监测30分钟内聚合情况,IC₅₀定义为抑制50%最大散射的浓度。FtsZ环破坏实验中,表达FtsZ-GFP的大肠杆菌用小檗碱(0–50 μM)处理2小时,甲醛固定后荧光显微镜观察FtsZ环形成[3]
蛋白质印迹和OPTDI分析用于检测细胞周期蛋白[1] 收获LoVo细胞,在100°C的裂解缓冲液[50 mmol/L TrisCl(pH 6.8)、100 mmol/L DTT、2%SDS、0.1%溴酚蓝、10%甘油]中裂解10分钟,并在−20°C下储存。蛋白质浓度通过BCA测定法测定。将等量的蛋白质装载到SDS聚丙烯酰胺凝胶上,并将蛋白质电泳转移到PVDF膜上。使用细胞周期蛋白B1、cdc2和cdc25c的特异性一级抗体(1:200稀释)分析免疫印迹,并与辣根过氧化物酶偶联的二级抗体(1:1000稀释)孵育,并使用增强化学发光检测试剂盒观察蛋白质。通过自动图像分析系统对光密度积分(OPTDI)进行分析。将细胞周期蛋白B1、cdc2和cdc25c的表达标准化为内部对照(GAPDH)。结果以处理与对照相比的百分比表示 DNA和蛋白质合成的测量[1] 通过3H-胸苷和L-[4,5-3H]-亮氨酸(分别为60 Ci/mg分子和0.5μCi/孔)的细胞掺入来评估DNA和蛋白质合成。将分离的细胞(每孔1×105个细胞)与含有一系列浓度的黄连素的培养基一起孵育。在24小时黄连素暴露前4小时,将放射性前体加入培养物中。在培养期结束时,将培养基移到一片滤膜上;用蒸馏水洗涤细胞三次。用液体闪烁光谱法测定3H-胸苷和L-[4,5-3H]-亮氨酸的掺入量。 |
| 细胞实验 |
细胞增殖实验[1]
细胞类型:四种结直肠癌细胞系LoVo、HCT116、SW480和HT-29 测试浓度:1.25、2.5、 5、10、20、40、80 和 160μM 孵育时间: 72 小时 实验结果: 抑制四种细胞的增殖细胞系。 IC50 范围为 40.8±4.1 μM (LoVo) 至 98.6±2.9 μM (HCT116)。 细胞增殖测定[1] 细胞类型: 结直肠癌细胞系 LoVo 测试浓度: 1.25、2.5、5、10 、20、40、80 和 160 μM 孵育时间:24、48、72 小时 实验结果:诱导时间和剂量 细胞生长依赖性抑制。 72 小时时,160.0 μM 在 LoVo 细胞中诱导 71.1±1.9% 的生长抑制。 细胞周期分析[1] 细胞类型: LoVo Cell 测试浓度: 0、10、20、40 或 80 μM 孵育持续时间:24 小时 实验结果:暴露于 40.0 μM 会诱导细胞周期停滞在 G2/M 期并增加 G2 /M期群体和G1期群体逐渐减少。 蛋白质印迹分析[1] 细胞类型: LoVo Cell 测试浓度: 10、20、40 或 80 μM 孵育持续时间:24小时 实验结果:循环抑制 1. 结直肠腺癌细胞实验(HCT116/SW480): - MTT实验:细胞(5×10³/孔)用含10%胎牛血清的RPMI 1640培养,小檗碱(10–200 μM)处理72小时,加入MTT(5 mg/mL)孵育4小时,DMSO裂解后测570 nm吸光度[1] - 凋亡实验:细胞(2×10⁵/孔)用小檗碱(25–100 μM)处理48小时,Annexin V-FITC/PI染色15分钟,流式细胞术分析[1] - Western blot:细胞用RIPA缓冲液裂解,30 μg蛋白SDS-PAGE分离,抗caspase-3/Bax/Bcl-2/GAPDH抗体孵育,ECL显色[1] 2. 多巴胺能神经元实验(SH-SY5Y): - 细胞活力实验:细胞(1×10⁴/孔)用小檗碱(1–20 μM)预处理2小时,再用6-OHDA(100 μM)处理24小时,MTT法测活力[5] - ROS检测:细胞负载DCFH-DA(10 μM)30分钟,小檗碱+6-OHDA处理后,测488/525 nm荧光[5] - HO-1 Western blot:细胞用小檗碱(5–20 μM)处理24小时,裂解后抗HO-1抗体检测[5] 3. 细菌FtsZ实验(大肠杆菌): - 表达FtsZ-GFP的大肠杆菌在LB培养基中培养,小檗碱(0–50 μM)处理2小时,4%多聚甲醛固定,共聚焦显微镜观察FtsZ环[3] |
| 动物实验 |
动物/疾病模型: 5周龄BALB/c nu/nu(裸鼠)人结直肠腺癌LoVo异种移植瘤[胃]与人结直肠腺癌裸鼠。异种移植瘤生长抑制率分别为33.1%和45.3%[1]。1]
剂量: 10、30或50 mg/kg/天 给药途径: 胃肠道灌胃; 10天 实验结果:在30和50 mg/kg/天的剂量下,抑制率分别为33.1%和45.3%。 小檗碱在人结直肠腺癌(LoVo)中的体内抗肿瘤作用[1] 采用裸鼠模型,以人结直肠腺癌LoVo异种移植瘤为模型,检测小檗碱的体内抗肿瘤功效;将1 × 10⁷个细胞皮下注射(sc)于5周龄BALB/c nu/nu小鼠的侧腹部。待肿瘤生长至约1,000–1,500 mm³后,处死小鼠并将肿瘤分割成等份。将6–8 mm³的结直肠腺癌组织碎片皮下植入5周龄BALB/c裸鼠的侧腹部。肿瘤生长2周后,将小鼠随机分为五组。小檗碱治疗组(每组10只小鼠)连续10天通过胃管灌注给予10、30或50 mg kg⁻¹ day⁻¹的小檗碱。5-氟尿嘧啶治疗组(每组10只小鼠)连续10天通过腹腔注射给予30 mg kg⁻¹ day⁻¹的5-氟尿嘧啶。对照组(每组11只小鼠)给予无菌水。每1–3天记录小鼠的体重和肿瘤体积,直至实验结束,此时肿瘤已严重影响小鼠的生存。测量肿瘤的长轴 (L) 和短轴 (S),并使用以下公式计算肿瘤体积 (V):V = S × S × L/2。完成最终测量后,通过颈椎脱臼处死小鼠。通过比较对照组和治疗组的肿瘤体积来确定抑制率:(1 − V治疗/V对照)。 小檗碱联合 5-氟尿嘧啶对裸鼠体内人结直肠腺癌 (HT-29) 异种移植瘤生长的影响[1] 本研究采用裸鼠人结直肠腺癌 HT-29 异种移植瘤模型,检测小檗碱联合 5-氟尿嘧啶的体内抗肿瘤疗效;将 1 × 107 个细胞皮下注射 (sc) 至 5 周龄 BALB/c nu/nu 小鼠的侧腹部。当肿瘤生长至约1000–1500 mm³时,处死小鼠并将肿瘤分割成大小相等的碎片。将6–8 mm³的结直肠腺癌碎片皮下植入5周龄BALB/c裸鼠的侧腹部。肿瘤生长3周后,将小鼠随机分为四组。小檗碱治疗组(10只小鼠)连续10天通过胃管灌注给予50 mg kg⁻¹ day⁻¹的小檗碱。5-氟尿嘧啶治疗组(10只小鼠)连续10天通过腹腔注射给予30 mg kg⁻¹ day⁻¹的5-氟尿嘧啶。联合治疗组(10只小鼠)同时给予小檗碱和5-氟尿嘧啶。对照组(10只小鼠)给予无菌水。每隔 3-4 天记录小鼠的体重和肿瘤体积,直至实验终点,此时肿瘤已严重影响小鼠的健康。测量肿瘤的长轴 (L) 和短轴 (S),并使用以下公式计算肿瘤体积 (V):V = S × S × L/2。完成最后一次测量后,通过颈椎脱臼处死小鼠。通过比较对照组和治疗组的肿瘤体积来确定抑制率:(1 − V治疗/V对照)。结直肠腺癌异种移植模型(裸鼠): - 动物:雄性裸鼠(6-8周龄,n=8/组)[1] - 肿瘤诱导:将5×10⁶个HCT116细胞(1:1 PBS:Matrigel)皮下植入右侧腹部[1] - 给药:将小檗碱溶于0.9%生理盐水中,腹腔注射,剂量分别为25/50/100 mg/kg,每日一次,连续21天;对照组注射生理盐水[1] - 评估:每周两次测量肿瘤体积(V=0.5×长×宽²);取出肿瘤进行称重和TUNEL染色[1] 2.生物利用度模型(Sprague-Dawley 大鼠): - 动物:雄性大鼠(200–220 g,每组 n=6)[2] - 给药:将柠檬酸/盐酸小檗碱溶于 0.5% CMC-Na 溶液中,以 100 mg/kg 的剂量口服给药;于 0.25–24 小时从眼眶静脉采集血液样本[2] - 评价:采用高效液相色谱法 (HPLC) 测定血浆小檗碱浓度;采用 DAS 软件计算药代动力学参数(Cmax、Tmax、t₁/₂、生物利用度)[2] |
| 药代性质 (ADME/PK) |
1. 小檗碱盐的口服生物利用度: - 在大鼠中,柠檬酸小檗碱(口服,100 mg/kg)的生物利用度(18.2±2.1%)高于盐酸小檗碱(6.5±0.8%)[2]
- Cmax:89.5±9.2 ng/mL(柠檬酸盐)vs. 32.1±3.5 ng/mL(盐酸盐);Tmax:1.5±0.2 小时(两者);半衰期:4.2±0.3 小时(柠檬酸盐)vs. 3.8±0.2 小时(盐酸盐)[2] - 血浆蛋白结合率:柠檬酸小檗碱为 85±3%(通过大鼠血浆超滤法测定)[2] 2. 临床药代动力学: - 在健康志愿者(n=12)中,口服小檗碱(500 mg,盐酸盐)显示 Cmax=28.6±3.1 ng/mL,Tmax=2.0±0.3 小时,t₁/₂=5.1±0.4 小时,口服生物利用度=5.2±0.6% [6] 吸收、分布和排泄 ...可能经皮肤吸收... /小檗碱/ |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 临床安全性(荟萃分析):- 在 27 项临床试验(n=2569)中,小檗碱(500–1500 mg/天,8–24 周)不良事件 (AE) 发生率较低(18.3% vs. 对照组的 12.5%)。常见不良事件为轻度胃肠道反应:腹泻 (6.2%)、恶心 (4.1%) 和腹部不适 (3.8%);未报告严重不良事件(例如肝肾毒性)[6]
2. 临床前毒性:- 在接受柠檬酸小檗碱(200 mg/kg/天,口服,持续4周)治疗的大鼠中,未观察到体重、肝功能(ALT/AST)或肾功能(BUN/Cr)的显著变化[2] 非人类毒性值 大鼠口服LD50大于1 g/kg 大鼠腹腔注射LD50 88.5 mg/kg 大鼠肌肉注射LD50 14.5 mg/kg |
| 参考文献 |
|
| 其他信息 |
1. 作用机制: - 抗肿瘤:通过激活 caspase-3 和 Bax/Bcl-2 失衡诱导结直肠腺癌细胞凋亡 [1]
- 抗菌:通过靶向 FtsZ 聚合和环化抑制细菌细胞分裂 [3] - 神经保护:通过上调 HO-1 和减少 ROS 保护多巴胺能神经元免受 6-OHDA 损伤 [5] - 代谢调节:通过上调 GLUT4 提高胰岛素敏感性,并通过抑制胆固醇合成降低血脂 [6] 2. 剂量优化原理: - 开发小檗碱有机酸盐(例如柠檬酸盐)旨在通过提高水溶性和减少肠道代谢来增强口服生物利用度(18.2% 对比盐酸盐的 6.5%)[2] 治疗用途 实验用途(兽医):口服硫酸小檗碱(350-700毫克/公斤)可有效治疗小鼠肠道念珠菌感染。 实验用途:局部应用黄连复合物(如小檗碱)的眼用溶液,可实现人眼瞳孔散大、局部麻醉和/或角膜上皮缺损的荧光染色和/或压平式眼压计的荧光测量。 /中国:既往用途/ 苦味健胃剂;抗菌剂;抗疟疾剂;解热药/小檗碱/ 对黏膜有轻微的局部麻醉作用。/小檗碱/ |
| 分子式 |
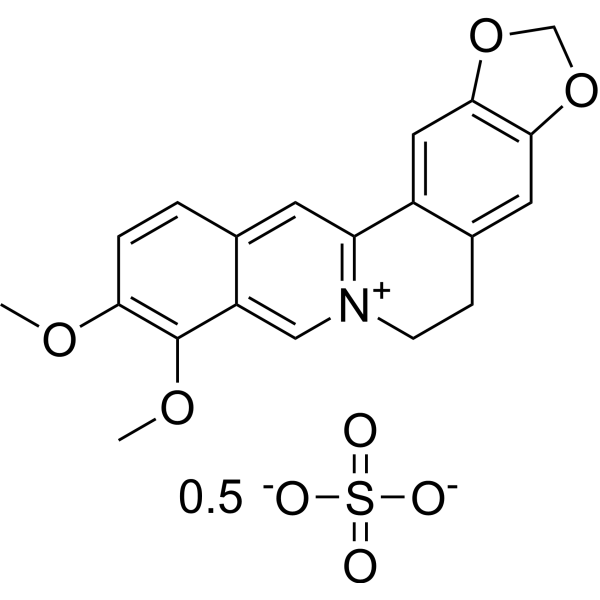
C20H18NO4.1/2O4S
|
|---|---|
| 分子量 |
384.40
|
| 精确质量 |
768.199
|
| CAS号 |
316-41-6
|
| 相关CAS号 |
2086-83-1 (cation);
117-74-8 (hydroxide); 1868138-66-2 (ursodeoxycholate); 633-65-8 (chloride); 633-66-9 (hydrosulfate); 316-41-6 (sulfate);
|
| PubChem CID |
9424
|
| 外观&性状 |
YELLOW CRYSTALS
|
| LogP |
5.935
|
| tPSA |
170.24
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
12
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
55
|
| 分子复杂度/Complexity |
551
|
| 定义原子立体中心数目 |
0
|
| SMILES |
COC1=C(C2=C[N+]3=C(C=C2C=C1)C4=CC5=C(C=C4CC3)OCO5)OC.COC1=C(C2=C[N+]3=C(C=C2C=C1)C4=CC5=C(C=C4CC3)OCO5)OC.O=S(=O)([O-])[O-]
|
| InChi Key |
JISRTQBQFQMSLG-UHFFFAOYSA-L
|
| InChi Code |
InChI=1S/C20H18NO4.H2O4S/c1-22-17-4-3-12-7-16-14-9-19-18(24-11-25-19)8-13(14)5-6-21(16)10-15(12)20(17)23-2;1-5(2,3)4/h3-4,7-10H,5-6,11H2,1-2H3;(H2,1,2,3,4)/q+1;/p-2
|
| 化学名 |
16,17-dimethoxy-5,7-dioxa-13-azoniapentacyclo[11.8.0.02,10.04,8.015,20]henicosa-1(13),2,4(8),9,14,16,18,20-octaene;sulfate
|
| 别名 |
Natural Yellow 18 hemisulfate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6015 mL | 13.0073 mL | 26.0146 mL | |
| 5 mM | 0.5203 mL | 2.6015 mL | 5.2029 mL | |
| 10 mM | 0.2601 mL | 1.3007 mL | 2.6015 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
