| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
EGFR
|
|---|---|
| 体外研究 (In Vitro) |
吉非替尼二盐酸盐(0.01–0.1 μM,72 小时)可增强贴壁依赖性生长和增殖,增强 ERK 信号传导,并增加受体的磷酸酪氨酸负载 [2]。使用吉非替尼二盐酸盐(1-2 μM,72 小时)观察到 EGFRvIII 磷酸酪氨酸负载、EGFRvIII 介导的增殖和锚定非依赖性生长显着降低 [2]。通过 STAT6 依赖性信号通路,吉非替尼二氯化物(0.62 μM,24-72 小时)抑制 IL-13 生成的 RAW 264.7 细胞的 M2 样极化 [3]。吉非替尼二氯化物(0.62 μM,72 小时)可抑制 M2 样巨噬细胞促进的侵袭和迁移 [3]。在 NSCLC 细胞系(H3255 和 HCC827 细胞)中,吉非替尼二氯化物(0–10 μM,72 小时)会导致细胞凋亡(诱导 BIM 蛋白)[4]。在 HCC827 和 A549 细胞中,gefitinib diHClide(100 nM,24 h)促进细胞外囊泡 (EV) 的吸收并抑制巨胞饮作用 [6]。顺铂耐药的 wtEGFR NSCLC 细胞系 H358R 和 A549R 的生长更容易受到吉非替尼二盐酸化物(1.5–60 μM,48 小时)的抑制[7]。
|
| 体内研究 (In Vivo) |
吉非替尼二盐酸盐(口服,75 mg/kg/d,21 天)可抑制 LLC 小鼠转移模型中巨噬细胞的 M2 样极化 [3]。吉非替尼二盐酸盐(口服,第一周 75 mg/kg,每天一次,每周 5 天)消除小叶增生和癌症中 HER2 和 HER3 的磷酸化以及通过 MAPK 和 Akt 的信号传导,增加脾细胞、淋巴中的 MAPK 活性和细胞因子产生节点[5]。吉非替尼二盐酸盐(150 mg/kg,口服,每日)可提高顺铂在 H358R 异种移植物中的抗癌活性 [7]。
|
| 细胞实验 |
蛋白质印迹分析[2]
细胞类型: NR6wtEGFR、NR6W 和 NR6M 测试浓度: 1、10、100 μM 孵育持续时间:5小时 实验结果:抑制EGFR酪氨酸磷酸化。 细胞迁移测定 [3] 细胞类型: LLC 细胞 测试浓度: 0.62 μM 孵育时间:72小时 实验结果:消除M2样巨噬细胞促进LLC的侵袭和迁移。 |
| 动物实验 |
动物/疾病模型: LLC小鼠转移模型[3]
剂量: 75 mg/kg/d,持续21天。 给药途径: 口服 实验结果: 减少肺转移结节数量,下调M2标志基因的表达以及肿瘤组织中CD206+和CD68+巨噬细胞的比例。 动物/疾病模型: BALB-NeuT转基因小鼠模型[5] 剂量: 第一周75 mg/kg,之后每隔一周增加15 mg/kg,每日一次,每周5天,之后停药2天,重复8-9周。 给药途径: 口服给药 实验结果: 经治疗的小鼠肿瘤数量从 9.6 减少到 0.58 (83%),小叶和小叶结节的数量和大小也减少了。 |
| 参考文献 |
[1]. Wakeling AE, et al. ZD1839: an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002 Oct 15;62(20):5749-54.
[2]. Pedersen MW, et al. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer. 2005 Oct 17;93(8):915-23. [3]. Muhammad Tariq, et al. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol Sin. 2017 Nov;38(11):1501-1511. [4]. Mark S Cragg, et al. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007 Oct;4(10):1681-89; discussion 1690. [5]. Marie P Piechocki, et al. Gefitinib prevents cancer progression in mice expressing the activated rat HER2/neu. Int J Cancer. 2008 Apr 15;122(8):1722-9. [6]. Tomoya Takenaka, et al. Effects of gefitinib treatment on cellular uptake of extracellular vesicles in EGFR-mutant non-small cell lung cancer cells. Int J Pharm. 2019 Dec 15;572:118762. [7]. Amin Li, et al. Gefitinib sensitization of cisplatin-resistant wild-type EGFR non-small cell lung cancer cells. J Cancer Res Clin Oncol. 2020 Jul;146(7):1737-1749. |
| 其他信息 |
表皮生长因子受体 (EGFR) 因其在肿瘤生长、转移和血管生成以及肿瘤对化疗和放疗的耐药性中发挥重要作用,而成为抗癌治疗的一个极具潜力的靶点。我们开发了一种低分子量 EGFR 酪氨酸激酶抑制剂 (EGFR-TKI) ZD1839(Iressa(2))。ZD1839 是一种取代的苯胺基喹唑啉类化合物,是一种强效的 EGFR-TKI (IC50 = 0.033 μM),可选择性抑制 EGF 刺激的肿瘤细胞生长 (IC50 = 0.054 μM),并阻断 EGF 刺激的肿瘤细胞中 EGFR 的自身磷酸化。在携带多种人源肿瘤异种移植瘤的小鼠模型中,每日一次口服 ZD1839 可剂量依赖性地抑制肿瘤生长。 EGFR的表达水平并不决定异种移植瘤对ZD1839的敏感性。对携带A431异种移植瘤的小鼠进行长期(>3个月)ZD1839治疗耐受性良好,ZD1839完全抑制了肿瘤生长并诱导了已形成肿瘤的消退。在ZD1839治疗期间未出现耐药性肿瘤,但部分肿瘤在停药后复发。这些研究表明ZD1839在治疗多种人类肿瘤方面具有潜在应用价值,并提示每日一次口服给药可能是一种合适的治疗方案。[1]表皮生长因子受体(EGFR)在多种人类肿瘤中经常发生扩增和/或突变,该受体的异常信号传导被认为与这些肿瘤的恶性表型有关。吉非替尼是一种小分子抑制剂,可特异性结合并抑制EGFR酪氨酸激酶,已被证实能够抑制多种EGFR过表达肿瘤细胞的生长、增殖、存活和侵袭。然而,吉非替尼的临床疗效与EGFR水平和活性之间缺乏相关性,表明其他分子机制,例如下游信号通路和基因突变,可能在预测临床疗效方面发挥重要作用。因此,我们研究了特异性EGFR抑制剂吉非替尼对表达天然存在的组成型活性EGFR变体EGFRvIII、低水平非转化EGFR和高水平转化EGFR的细胞的磷酸化水平、信号通路和生长的影响。结果表明,足以抑制EGFR磷酸化、EGFR介导的增殖和EGFR介导的非锚定依赖性生长的吉非替尼剂量,不足以抑制表达EGFRvIII细胞的这些特征。此外,数据表明,EGFRvIII 表达细胞长期暴露于低浓度吉非替尼(0.01-0.1 μM)会导致受体磷酸酪氨酸水平升高,ERK 信号通路增强,并刺激细胞增殖和非锚定依赖性生长,这可能是通过诱导 EGFRvIII 二聚化实现的。另一方面,较高浓度的吉非替尼(1-2 μM)则显著降低 EGFRvIII 磷酸酪氨酸水平、EGFRvIII 介导的细胞增殖和非锚定依赖性生长。需要进一步研究以探讨这些重要发现的临床意义。[2] M2 样极化肿瘤相关巨噬细胞 (TAM) 在促进癌细胞生长、侵袭、转移和血管生成中发挥着关键作用。在肿瘤进展过程中识别 M2 样 TAM 是癌症治疗的一种有吸引力的策略。本研究在体外和体内探讨了巨噬细胞极化与吉非替尼抗肿瘤作用在Lewis肺癌(LLC)中的作用。浓度低于2.5 μmol/L的吉非替尼对LLC和RAW 264.7细胞系以及骨髓来源巨噬细胞(BMDM)的生长没有显著抑制作用。然而,低浓度吉非替尼(0.62 μmol/L)显著抑制了IL-13诱导的巨噬细胞M2样极化,表现为M2表面标志物CD206和CD163表达降低、特定M2标志基因(Mrc1、Ym1、Fizz1、Arg1、IL-10和CCL2)表达下调,以及M2样巨噬细胞介导的LLC细胞侵袭和迁移受到抑制。在RAW 264.7细胞中,吉非替尼抑制IL-13诱导的STAT6磷酸化,而STAT6磷酸化是巨噬细胞M2样极化过程中的关键信号通路。在LLC小鼠转移模型中,口服吉非替尼(75 mg·kg-1·d-1,连续21天)显著减少了肺转移结节的数量,下调了肿瘤组织中M2标志基因的表达以及CD206+和CD68+巨噬细胞的比例。这些结果表明,吉非替尼在体外和体内均能有效抑制M2样极化,揭示了吉非替尼化学预防作用的一种新的潜在机制。[3]
|
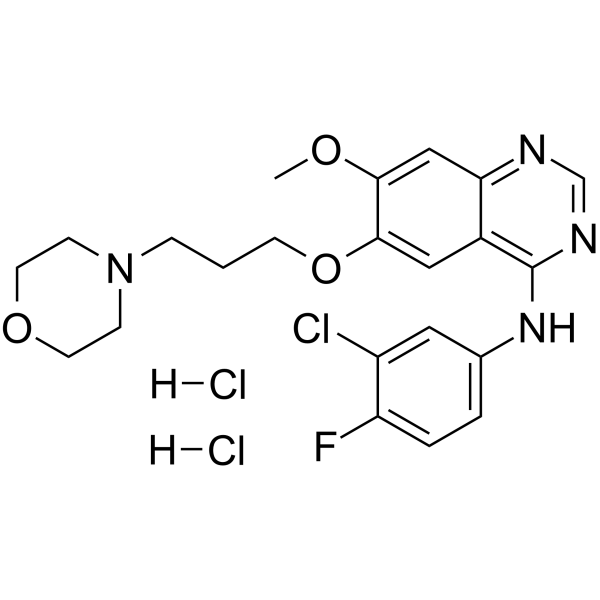
| 分子式 |
C22H24N4O3FCL.2[HCL]
|
|---|---|
| 分子量 |
519.82424
|
| 精确质量 |
518.105
|
| CAS号 |
184475-56-7
|
| 相关CAS号 |
Gefitinib;184475-35-2
|
| PubChem CID |
19077503
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 沸点 |
628.2ºC at 760 mmHg
|
| 闪点 |
333.7ºC
|
| 蒸汽压 |
4.9E-16mmHg at 25°C
|
| LogP |
5.89
|
| tPSA |
68.74
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
545
|
| 定义原子立体中心数目 |
0
|
| SMILES |
COC1=C(C=C2C(=C1)N=CN=C2NC3=CC(=C(C=C3)F)Cl)OCCCN4CCOCC4.Cl
|
| InChi Key |
OHAYARNLYSPHOJ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C22H24ClFN4O3.2ClH/c1-29-20-13-19-16(12-21(20)31-8-2-5-28-6-9-30-10-7-28)22(26-14-25-19)27-15-3-4-18(24)17(23)11-15;;/h3-4,11-14H,2,5-10H2,1H3,(H,25,26,27);2*1H
|
| 化学名 |
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine;dihydrochloride
|
| 别名 |
184475-56-7; Gefitinib (Dihydrochloride); 4-(3-Chloro-4-fluorophenylamino)-7-methoxy-6-[3-(4-morpholinyl)propoxy]quinazoline dihydrochloride; Gefitinib 2hydrochloride salt; gefitinib dihydrochloride; N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine;dihydrochloride; ZD 1839 Dihydrochloride;ZD-1839 Dihydrochloride;ZD1839 Dihydrochloride; SCHEMBL8208642;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9237 mL | 9.6187 mL | 19.2374 mL | |
| 5 mM | 0.3847 mL | 1.9237 mL | 3.8475 mL | |
| 10 mM | 0.1924 mL | 0.9619 mL | 1.9237 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03292133 | Active Recruiting |
Drug: EGF816 Drug: Gefitinib |
Lung Cancer | Massachusetts General Hospital | October 31, 2017 | Phase 2 |
| NCT03122717 | Active Recruiting |
Drug: Gefitinib Drug: Osimertinib |
Non-Small Cell Lung Cancer | Dana-Farber Cancer Institute | May 9, 2017 | Phase 1 Phase 2 |
| NCT03758287 | Active Recruiting |
Drug: Gefitinib Drug: CT053PTSA |
Non-small Cell Lung Cancer | Sunshine Lake Pharma Co., Ltd. | November 2016 | Phase 1 Phase 2 |
| NCT03849768 | Active Recruiting |
Drug: Gefitinib Drug: HS-10296 |
Non Small Cell Lung Cancer | Jiangsu Hansoh Pharmaceutical Co., Ltd. |
February 1, 2019 | Phase 3 |
| NCT02856893 | Active Recruiting |
Drug: Gefitinib Drug: Osimertinib |
NSCLC | European Organisation for Research and Treatment of Cancer - EORTC |
October 10, 2017 | Phase 2 |
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
