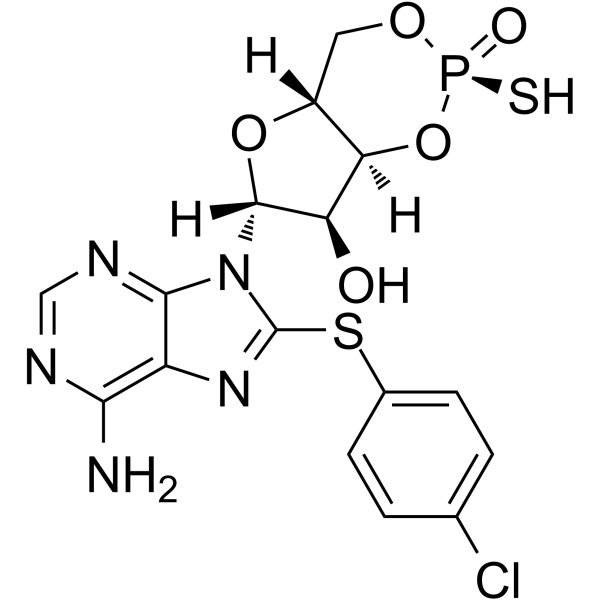
| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
PKA[1]
|
|---|---|
| 体外研究 (In Vitro) |
Rp-8-CPT-cAMPS(100 μM;15 分钟)可显着降低毛喉素和非诺特罗引起的 VASP 磷酸化,并抑制 6-Bnz-cAMP 引起的 VASP 磷酸化[2]。 Rp-8-CPT-cAMPS(100 μM;30 分钟)通过 8-pCPT-2'-O-Me-cAMP 和 6-Bnz-cAMP 降低 Rap1 的 GTP 负载量[2]。 Rp-8-CPT-cAMPS (100 μM; 30 分钟)[2]。在预收缩的大鼠肠系膜动脉环中,Rp-8-CPT-cAMPS (10 μM) 可减少毒液引起的内皮依赖性和非依赖性松弛[3]。
|
| 细胞实验 |
在基础条件下、单独或联合非诺特罗刺激、Epac激活剂8-pCPT-2′-O-Me-cAMP和Sp-8-pCPT-2′-O-Me-cAMP、PKA激活剂6-Bnz-cAMP和cGMP类似物8-pCPT-2′-O-Me-cGMP后,通过ELISA法评估IL-8的释放。在有指示的情况下,用药物抑制剂艰难梭菌毒素B-1470 (GTPases)、U0126(细胞外信号调节激酶ERK1/2)和rp -8- cpt - camp (PKA)对细胞进行预孵育。通过测量PKA底物血管扩张剂刺激磷酸化蛋白的磷酸化,证实了环核苷酸类似物的特异性。采用下拉法测定Rap1和Rap2的gtp负载。western blot检测Rap1、Rap2、Epac1和Epac2的表达。通过siRNA实现Epac蛋白表达下调。采用非配对或配对双尾Student's t检验[2]。
|
| 参考文献 |
|
| 其他信息 |
series of cAMP analogs were synthesized by introducing exocyclic sulfur substitutions at the equatorial (Rp) or axial (Sp) positions of the cyclic phosphate ring and modifying the adenine bases of cAMP. The ability of these compounds to inhibit the binding of [3H]cAMP to the A and B sites of type I (rabbit skeletal muscle) and type II (bovine cardiac muscle) cAMP-dependent protein kinases was quantitatively determined. On average, the Sp isomer showed a 5-fold lower affinity for the A site of the type I isoenzyme and a 30-fold lower affinity for the B site compared to its cyclic phosphate homologue. Affinities for the corresponding sites of the type II isoenzymes were reduced by an average of 20-fold and 4-fold, respectively. Compared to the A and B sites of isoenzyme II, the Rp isomer showed approximately 400-fold and 200-fold decreased affinity for the A and B sites of isoenzyme I, respectively, while its affinity for the A and B sites of isoenzyme II decreased by approximately 200-fold and 45-fold, respectively. Therefore, Sp substitution increased the relative preference for the A site of isoenzyme I and the B site of isoenzyme II. On the other hand, Rp substitution increased the relative preference for the B sites of both isoenzymes. These data suggest that the two intrachain sites of isoenzymes I and II exhibit different tolerances to Rp and Sp substitutions. They also support the hypothesis that, in all four binding sites, the negative charge interacting with conserved arginine is provided by the axial oxygen atom (rather than the previously proposed equatorial oxygen atom). Furthermore, they demonstrated that the combined modification of the cAMP adenine ring and the cyclic phosphate ring enhances the ability to distinguish between the A and B sites of isoenzymes and between isoenzymes I and II. Since cAMP Rp analogs are known to inhibit the activation of cAMP-dependent protein kinases, the findings of this study are of great significance for the synthesis of analogs with high selectivity for isoenzyme I or II. [1]
Background: Airway smooth muscle participates in the pathogenesis of lung diseases by secreting inflammatory mediators such as interleukin-8 (IL-8). The production of IL-8 is partially regulated by the activation of Gq and Gs coupled receptors. This study investigated the role of cyclic adenosine monophosphate (cAMP) effector protein kinase A (PKA) and cAMP-directly activated exchange proteins (Epac1 and Epac2) in bradykinin-induced release of IL-8 in human airway smooth muscle cell lines and their potential molecular mechanisms. [2] Results: The β2 receptor agonist fennotefurano enhanced bradykinin-induced IL-8 release. PKA activator 6-Bnz-cAMP and Epac activator 8-pCPT-2'-O-Me-cAMP significantly increased bradykinin-induced IL-8 release. The hydrolysis-resistant Epac activator Sp-8-pCPT-2'-O-Me-cAMPS mimicked the effect of 8-pCPT-2'-O-Me-cAMP, while the negative control 8-pCPT-2'-O-Me-cGMP did not. Fenoterol, foskeline, and 6-Bnz-cAMP induced VASP phosphorylation, while the PKA inhibitor Rp-8-CPT-cAMPS attenuated this phosphorylation. 6-Bnz-cAMP and 8-pCPT-2'-O-Me-cAMP induced GTP binding of Rap1, but had no effect on Rap2. Treatment of cells with toxins B-1470 and U0126 significantly reduced bradykinin-induced IL-8 release, whether used alone or in combination with PKA and Epac activators. Interestingly, the inhibition of PKA by Rp-8-CPT-cAMPS and the silencing of Epac1 and Epac2 expression by specific siRNAs significantly reduced Rap1 activation and the enhancing effect of PKA and Epac on bradykinin-induced IL-8 release. [2] Conclusion: In summary, our data suggest that PKA, Epac1, and Epac2 work synergistically to regulate the inflammatory properties of airway smooth muscle by signaling to Ras-like GTPases Rap1 and ERK1/2. [2] Sudden death following venom poisoning in some Australian cobra species is a cause of death that is not fully understood. We have previously demonstrated that the venom of Oxyuranus scutellatus causes cardiovascular failure in anesthetized rats. Pre-administration of a sublethal dose of venom attenuated the response to subsequent administration of a higher (lethal) dose. This study explored the possible mechanisms mediating this "protective effect." Baboon apanzan venom (5 μg/kg, intravenously) induced mild, transient hypotension in anesthetized rats, while a dose of 10 μg/kg resulted in a 73 ± 12% decrease in arterial blood pressure. The venom (20 μg/kg or 50 μg/kg) induced cardiovascular failure in all tested animals (n=12). Pre-administration of "starting" doses of venom (5, 10, and 20 μg/kg) prevented cardiovascular failure induced by 50 μg/kg venom. Furthermore, pre-administration of indomethacin (30 mg/kg) or heparin (300 units/kg) also prevented sudden death induced by venom (20 μg/kg). The venom had no effect on isolated hearts, suggesting that direct cardiac effects are unlikely to be the cause of the "sudden death." The venom induced endothelium-dependent and non-endothelium-dependent relaxation of preconstricted rat mesenteric arterial rings, which was inhibited by indomethacin, IbTx, and Rp-8-CPT-cAMPs. This relaxation was significantly reduced after a second exposure to the venom. Our results suggest that cardiovascular failure induced by O. scutellatus venom may be the result of the combined effects of the release of multiple substances. [3] |
| 分子式 |
C16H15CLN5O5PS2
|
|---|---|
| 分子量 |
487.88
|
| 精确质量 |
508.976
|
| CAS号 |
129735-01-9
|
| 相关CAS号 |
Rp-8-CPT-cAMPS sodium;221905-35-7;Sp-8-CPT-cAMPS;129693-13-6
|
| PubChem CID |
23679060
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
3.773
|
| tPSA |
207.8
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
11
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
701
|
| 定义原子立体中心数目 |
4
|
| SMILES |
[Na+].ClC1C=CC(SC2=NC3C(=NC=NC=3N2[C@@H]2O[C@@H]3COP(O[C@H]3[C@H]2O)([O-])=S)N)=CC=1
|
| InChi Key |
AWXMSJRRXLCVMW-JVSRKQJHSA-M
|
| InChi Code |
InChI=1S/C16H15ClN5O5PS2.Na/c17-7-1-3-8(4-2-7)30-16-21-10-13(18)19-6-20-14(10)22(16)15-11(23)12-9(26-15)5-25-28(24,29)27-12;/h1-4,6,9,11-12,15,23H,5H2,(H,24,29)(H2,18,19,20);/q;+1/p-1/t9-,11-,12-,15-,28?;/m1./s1
|
| 化学名 |
sodium;(4aR,6R,7R,7aS)-6-[6-amino-8-(4-chlorophenyl)sulfanylpurin-9-yl]-2-oxido-2-sulfanylidene-4a,6,7,7a-tetrahydro-4H-furo[3,2-d][1,3,2]dioxaphosphinin-7-ol
|
| 别名 |
Rp-8-CPT-cAMPS; 129735-01-9; Sp-8-pCPT-cAMPS; 221905-35-7; Rp-8-CPT-CyclicAMP(SodiumSalt); Rp-8-CPT-cAMPS sodium; Rp-8-CPT-cAMPS (sodium); Rp-8-CPT-Cyclic AMP (sodium salt);
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0497 mL | 10.2484 mL | 20.4968 mL | |
| 5 mM | 0.4099 mL | 2.0497 mL | 4.0994 mL | |
| 10 mM | 0.2050 mL | 1.0248 mL | 2.0497 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Kiss2 peptide acetate
Kiss2 peptide acetate
 Rp-8-Br-cAMPS sodium
Rp-8-Br-cAMPS sodium
 8-Bromo-cAMP
8-Bromo-cAMP
 Sp-8-Br-cAMPS
Sp-8-Br-cAMPS
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
