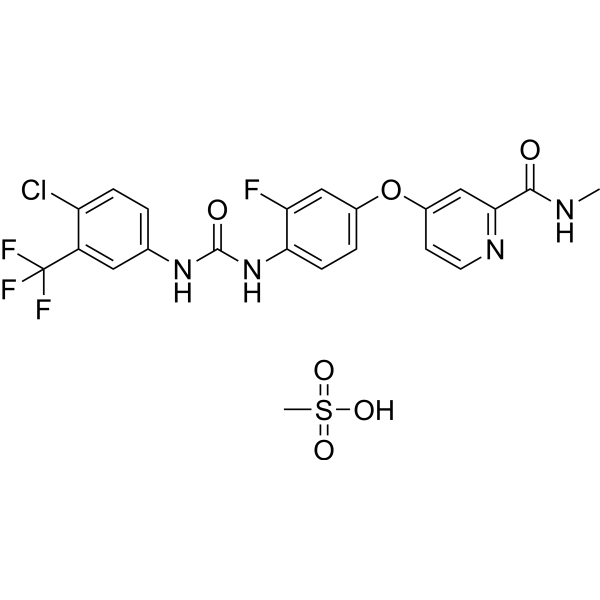
Regorafenib (BAY 73-4506) 甲磺酸盐是一种口服生物可利用的多靶点受体酪氨酸激酶(酪氨酸激酶)抑制剂,分别抑制 VEGFR1/2/3、PDGFRβ、Kit、RET 和 Raf-1 的 IC50。 13/4.2/46、22、7、1.5 和 2.5 nM。甲磺酸瑞戈非尼显示出非常有效的抗肿瘤和抗血管生成活性。
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
A Trial to Evaluate Multiple Regimens in Newly Diagnosed and Recurrent Glioblastoma
CTID: NCT03970447
Phase: Phase 2/Phase 3 Status: Recruiting
Date: 2024-12-02
Regorafenib for Recurrent Grade 2 and 3 Meningioma (MIRAGE Trial)
CTID: NCT06275919
Phase: Phase 2 Status: Recruiting
Date: 2024-11-29
Phase Ib / Regorafenib With Conventional Chemotherapy/Newly Diagnosed Patients/ Multimetastatic Ewing Sarcoma
CTID: NCT05830084
Phase: Phase 1 Status: Recruiting
Date: 2024-11-29
An Open Label Study Evaluating the Efficacy and Safety of Etrumadenant (AB928) Based Treatment Combinations in Participants With Metastatic Colorectal Cancer.
CTID: NCT04660812
Phase: Phase 1/Phase 2 Status: Active, not recruiting
Date: 2024-11-25
Botensilimab, Balstilimab and Regorafenib or Botensilimab and Balstilimab for the Treatment of Advanced or Metastatic Microsatellite Stable Colorectal Cancer
CTID: NCT06575725
Phase: Phase 2 Status: Withdrawn
Date: 2024-11-21
View More
A Study of Continued Treatment With Regorafenib in Participants With Solid Tumors Who Have Participated in Other Bayer Studies
CTID: NCT06246643
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-11-20
Evaluation of Treatment PERSOnalization Based on Its Therapeutic Monitoring in Patients with Metastatic Colorectal Cancer Treated with REgorafenib
CTID: NCT04874207
Phase: Phase 4 Status: Active, not recruiting
Date: 2024-11-15
Regorafenib in Patients with Progressive, Recurrent/Metastatic Adenoid Cystic Carcinoma
CTID: NCT02098538
Phase: Phase 2 Status: Completed
Date: 2024-11-12
Safety and Efficacy of SYHA1813 Single Agent or in Combination With Different Regimens in Unresectable Locally Advanced or Metastatic Solid Tumors.
CTID: NCT06682611
Phase: Phase 1/Phase 2 Status: Not yet recruiting
Date: 2024-11-12
A Study Using Regorafenib as Second or Third Line Therapy in Metastatic Medullary Thyroid Cancer
CTID: NCT02657551
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-11-12
TAPUR: Testing the Use of Food and Drug Administration (FDA) Approved Drugs That Target a Specific Abnormality in a Tumor Gene in People With Advanced Stage Cancer
CTID: NCT02693535
Phase: Phase 2 Status: Recruiting
Date: 2024-11-12
An Observational Study Called STAR-T to Learn More About the Sequential Treatment With Regorafenib and TAS-102 in Adults With Metastatic Colorectal Cancer Under Real World Conditions
CTID: NCT05839951
Phase: Status: Active, not recruiting
Date: 2024-11-12
An Open-Label Study to Enable Continued Treatment Access for Subjects Previously Enrolled in Studies of Ruxolitinib
CTID: NCT02955940
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-11-06
A Real-World Study to Learn More About the Order of Different Treatments and Their Effects in People With Metastatic Colorectal Cancer Receiving Their Third and Fourth Line of Treatment
CTID: NCT06137170
Phase: Status: Active, not recruiting
Date: 2024-11-01
A Study of Coformulated Favezelimab/Pembrolizumab (MK-4280A) Versus Standard of Care in Subjects With Previously Treated Metastatic PD-L1 Positive Colorectal Cancer (MK-4280A-007)-China Extension Study
CTID: NCT05600309
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-10-30
A Study of Coformulated Favezelimab/Pembrolizumab (MK-4280A) Versus Standard of Care in Subjects With Previously Treated Metastatic PD-L1 Positive Colorectal Cancer (MK-4280A-007)
CTID: NCT05064059
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-10-30
Study of Lenvatinib (MK-7902/E7080) in Combination With Pembrolizumab (MK-3475) Versus Standard of Care in Participants With Metastatic Colorectal Cancer (MK-7902-017/E7080-G000-325/LEAP-017)
CTID: NCT04776148
Phase: Phase 3 Status: Completed
Date: 2024-10-29
ctDNA-Guided Sunitinib And Regorafenib Therapy for GIST
CTID: NCT05366816
Phase: Phase 2 Status: Recruiting
Date: 2024-10-26
Safety and Efficacy of NEO212 in Patients with Astrocytoma IDH-mutant, Glioblastoma IDH-wildtype or Brain Metastasis
CTID: NCT06047379
Phase: Phase 1/Phase 2 Status: Recruiting
Date: 2024-10-16
A Study Evaluating the Activity of Anti-cancer Treatments Targeting Tumor Molecular Alterations/characteristics in Advanced / Metastatic Tumors.
CTID: NCT04116541
Phase: Phase 2 Status: Recruiting
Date: 2024-10-16
An Observational Study to Learn More About the Long-Term Responses to Treatment With Regorafenib in Patients With Metastatic Colorectal Cancer in the United States
CTID: NCT06029010
Phase: Status: Active, not recruiting
Date: 2024-10-15
A Phase 2 Randomized, Open-Label Study of RRx-001 vs Regorafenib in Subjects With Metastatic Colorectal Cancer
CTID: NCT02096354
Phase: Phase 2 Status: Completed
Date: 2024-10-15
Sotorasib and Panitumumab Versus Investigator's Choice for Participants With Kirsten Rat Sarcoma (KRAS) p.G12C Mutation
CTID: NCT05198934
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-10-15
Regorafenib and Pembrolizumab in Treating Participants With Advanced or Metastatic Colorectal Cancer
CTID: NCT03657641
Phase: Phase 1/Phase 2 Status: Active, not recruiting
Date: 2024-10-10
An Investigational Immunotherapy Study of BMS-986288 Alone and in Combination With Nivolumab in Advanced Solid Cancers
CTID: NCT03994601
Phase: Phase 1/Phase 2 Status: Completed
Date: 2024-10-08
[18F]FLT-PET as a Predictive Imaging Biomaker of Treatment Responses to Regorafenib
CTID: NCT02175095
Phase: N/A Status: Completed
Date: 2024-10-04
Regorafenib in Combination with Pembrolizumab or Pembrolizumab for MSI-H Colorectal Cancer
CTID: NCT06006923
Phase: Phase 2 Status: Recruiting
Date: 2024-10-02
Regorafenib, With Cetuximab or Panitumumab, for the Treatment of Unresectable, Locally Advanced, or Metastatic Colorectal Cancer
CTID: NCT04117945
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-09-27
Botensilimab, Balstilimab and Regorafenib for the Treatment of Patients with Microsatellite Stable Metastatic Colorectal Cancer Who Have Progressed on Prior Chemotherapy
CTID: NCT05672316
Phase: Phase 1/Phase 2 Status: Active, not recruiting
Date: 2024-09-25
Regorafenib Alone or in Combination With Hypofractionated/Low-dose Radiotherapy Plus Toripalimab for Metastatic Colorectal Cancer
CTID: NCT05963490
Phase: Phase 2 Status: Recruiting
Date: 2024-09-24
Assessing a Regorafenib-irinotecan Combination Versus Regorafenib Alone in Metastatic Colorectal Cancer Patients
CTID: NCT03829462
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-09-20
Regorafenib and Nivolumab in Mismatch Repair (MMR) Refractory Colorectal Cancer
CTID: NCT03712943
Phase: Phase 1 Status: Completed
Date: 2024-09-19
Study Of Intrabucally Administered Electromagnetic Fields and Regorafenib
CTID: NCT04327700
Phase: Phase 2 Status: Terminated
Date: 2024-09-19
Regorafenib in Metastatic Colorectal Cancer
CTID: NCT02466009
Phase: Phase 2 Status: Completed
Date: 2024-09-04
Regorafenib Followed by Nivolumab in Patients With Hepatocellular Carcinoma (GOING)
CTID: NCT04170556
Phase: Phase 1/Phase 2 Status: Completed
Date: 2024-09-03
GSL Synthetase Inhibitor Plus Immune Checkpoint Inhibitor and/or Regorafenib in Previously Treated pMMR/MSS CRC.
CTID: NCT06558773
Phase: Phase 2 Status: Not yet recruiting
Date: 2024-08-21
An Investigational Immuno-therapy Study Of Nivolumab In Combination With Trametinib With Or Without Ipilimumab In Participants With Previously Treated Cancer of the Colon or Rectum That Has Spread
CTID: NCT03377361
Phase: Phase 1/Phase 2 Status: Active, not recruiting
Date: 2024-08-19
Neoadjuvant Regorafenib in Combination With Nivolumab and Short-course Radiotherapy in Stage II-III Rectal Cancer
CTID: NCT04503694
Phase: Phase 2 Status: Recruiting
Date: 2024-08-13
Regorafenib, Ipilimumab and Nivolumab for the Treatment of Chemotherapy Resistant Microsatellite Stable Metastatic Colorectal Cancer
CTID: NCT04362839
Phase: Phase 1 Status: Active, not recruiting
Date: 2024-08-05
Regorafenib and Durvalumab for the Treatment of High-Risk Liver Cancer
CTID: NCT05194293
Phase: Phase 2 Status: Recruiting
Date: 2024-07-30
Cadonilimab Combined With Regorafenib as A Third-line Treatment in Patients With MSS CRLM
CTID: NCT06455254
Phase: Phase 2 Status: Recruiting
Date: 2024-07-30
A Trial to Learn Whether Regorafenib in Combination With Nivolumab Can Improve Tumor Responses and How Safe it is for Participants With Solid Tumors
CTID: NCT04704154
Phase: Phase 2 Status: Completed
Date: 2024-07-23
The Finnish National Study to Facilitate Patient Access to Targeted Anti-cancer Drugs
CTID: NCT05159245
Phase: Phase 2 Status: Recruiting
Date: 2024-07-15
Clinical Study of Regorafenib and Nivolumab Plus Chemotherapy
CTID: NCT05394740
Phase: Phase 1/Phase 2 Status: Active, not recruiting
Date: 2024-07-08
Study of XL092 + Atezolizumab vs Regorafenib in Subjects With Metastatic Colorectal Cancer
CTID: NCT05425940
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-07-05
Circulating Cell-Free Tumor DNA Testing in Guiding Treatment for Patients With Advanced or Metastatic Colorectal Cancer
CTID: NCT03844620
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-06-28
Efficacy of Ginseng for Patients on Regorafenib
CTID: NCT02581059
Phase: Phase 2 Status: Terminated
Date: 2024-06-26
Phase 2 Study to Evaluate the Efficacy of Regorafenib in Specific GIST Mutation Subsets (KIT Exon 17, 18, or 14 Mutation and SDHB Deficient GIST) in the Post-imatinib Second-line Setting.
CTID: NCT06087263
Phase: Phase 2 Status: Recruiting
Date: 2024-06-21
Combined TACE, TKI/Anti-VEGF and ICIs as Conversion Therapy for Advanced Hepatocellular Carcinoma
CTID: NCT05717738
Phase: Status: Recruiting
Date: 2024-06-13
Regorafenib in Combination With Venetoclax and Azacitidine for the Treatment of Patients With Relapsed or Refractory Acute Myeloid Leukemia
CTID: NCT06454409
Phase: Phase 1 Status: Not yet recruiting
Date: 2024-06-12
Phase II Study of Regorafenib as Maintenance Therapy
CTID: NCT03793361
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-06-04
An Observational Study to Learn More About Treatment With Regorafenib in People With Advanced Gastrointestinal Stromal Tumors in the United States
CTID: NCT06321055
Phase: Status: Completed
Date: 2024-05-31
Regorafenib in Combination With Multimodal Metronomic Chemotherapy for Chemo-resistant Metastatic Colorectal Cancers
CTID: NCT06425133
Phase: Phase 2 Status: Not yet recruiting
Date: 2024-05-24
FaR-RMS: An Overarching Study for Children and Adults With Frontline and Relapsed RhabdoMyoSarcoma
CTID: NCT04625907
Phase: Phase 1/Phase 2 Status: Recruiting
Date: 2024-05-23
Panitumumab, Regorafenib, or TAS-102, in Treating Patients With Metastatic and/or Unresectable RAS Wild-Type Colorectal Cancer
CTID: NCT03992456
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-05-21
RegoNivo vs Standard of Care Chemotherapy in AGOC
CTID: NCT04879368
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-05-16
T-Cell Therapy (ECT204) in Adults With Advanced HCC
CTID: NCT04864054
Phase: Phase 2 Status: Recruiting
Date: 2024-05-06
Regorafenib Plus Pembrolizumab in Patients With Advanced or Spreading Liver Cancer Who Have Been Previously Treated With PD-1/PD-L1 Immune Checkpoint Inhibitors
CTID: NCT04696055
Phase: Phase 2 Status: Completed
Date: 2024-05-01
Regorafenib and XmAb20717 in Treatment of High-risk Patients With Colorectal Cancer With Radiographic Occult Molecular Residual Disease After End of Established Definitive Therapy (RX-CROME)
CTID: NCT05900648
Phase: Phase 2 Status: Withdrawn
Date: 2024-04-29
A Phase I Dose Finding Study in Children With Solid Tumors Recurrent or Refractory to Standard Therapy
CTID: NCT02085148
Phase: Phase 1 Status: Completed
Date: 2024-04-22
Regorafenib Combined With Fulvestrant in Recurrent Low-Grade Serous Ovarian Cancer
CTID: NCT05113368
Phase: Phase 2 Status: Recruiting
Date: 2024-04-11
A Clinical Study of Regorafenib in Participants Who Have Been Treated in Previous Bayer-sponsored Regorafenib Studies That Have Been Completed
CTID: NCT03890731
Phase: Phase 2 Status: Completed
Date: 2024-04-02
HAIC Combined With Cadonilimab and Regorafenib as 2nd-line Treatment for ICC
CTID: NCT06335927
Phase: Phase 2 Status: Recruiting
Date: 2024-03-28
Combination of LTC004 and Regorafenib to Treat Patients With Advanced/Metastatic CRC
CTID: NCT06322563
Phase: Phase 2 Status: Not yet recruiting
Date: 2024-03-21
A Study of Nivolumab Combined With FOLFOX and Regorafenib in People Who Have HER2-Negative Esophagogastric Cancer
CTID: NCT04757363
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-03-19
Study of Regorafenib in Patients With Advanced Myeloid Malignancies
CTID: NCT03042689
Phase: Phase 1 Status: Completed
Date: 2024-03-08
Serial Measurements of Molecular and Architectural Responses to Therapy (SMMART) PRIME Trial
CTID: NCT03878524
Phase: Phase 1 Status: Terminated
Date: 2024-03-04
A Study of Nivolumab-relatlimab Fixed-dose Combination Versus Regorafenib or TAS-102 in Participants With Later-lines of Metastatic Colorectal Cancer
CTID: NCT05328908
Phase: Phase 3 Status: Active, not recruiting
Date: 2024-03-01
A Clinical Study of BioTTT001 in Combination With Toripalimab and Regorafenib in Patients With Colorectal Cancer
CTID: NCT06283134
Phase: Phase 1 Status: Not yet recruiting
Date: 2024-02-29
A Clinical Study of T3011 in Combination With Toripalimab and Regorafenib in Patients With Colorectal Cancer
CTID: NCT06283303
Phase: Phase 1 Status: Not yet recruiting
Date: 2024-02-29
A Trial of Cadonilimab Plus Regorafenib in Patients With Hepatocellular Carcinoma Who Failed Camrelizumab Combined With Apatinib
CTID: NCT06280105
Phase: Phase 2 Status: Not yet recruiting
Date: 2024-02-28
Immune Checkpoint Therapy vs Target Therapy in Reducing Serum HBsAg Levels in Patients With HBsAg+ Advanced Stage HCC
CTID: NCT03899428
Phase: Phase 2 Status: Recruiting
Date: 2024-02-28
Chidamide + Regorafenib in Hepatocellular Carcinoma (HCC)
CTID: NCT05770882
Phase: Phase 1/Phase 2 Status: Recruiting
Date: 2024-02-23
Efficacy and the Safety of Regorafenib in Patients Aged More Than 70 Years With a Metastatic Colorectal Adenocarcinoma .
CTID: NCT02788006
Phase: Phase 2 Status: Completed
Date: 2024-02-23
The Purpose of This Trial is to Determine if Regorafenib Plus Durvalumab (MEDI4736) is Safe and Effective in Treatment of Chemo Refractory Advanced Biliary Tract Cancers
CTID: NCT04781192
Phase: Phase 1/Phase 2 Status: Recruiting
Date: 2024-02-23
Regorafenib With Temozolomide With or Without RT in MGMT-Methylated, IDH Wild-type GBM Patients
CTID: NCT06095375
Phase: Phase 1 Status: Recruiting
Date: 2024-02-12
Regorafenib in Patients With Relapsed Glioblastoma. IOV-GB-1-2020 REGOMA-OSS
CTID: NCT04810182
Phase: Status: Completed
Date: 2024-02-08
The Drug Rediscovery Protocol (DRUP Trial)
CTID: NCT02925234
Phase: Phase 2 Status: Recruiting
Date: 2024-01-24
Regorafenib in Good Performance Status Patients With Newly Diagnosed Metastatic Colorectal Adenocarcinoma
CTID: NCT02023333
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-01-22
Regorafenib in Patients With Refractory Primary Bone
A Phase 3 Multicenter, Randomized, Open-label, Active-controlled Study of Sotorasib and Panitumumab Versus Investigator’s Choice (Trifluridine and Tipiracil, or Regorafenib) for the Treatment of Previously Treated Metastatic Colorectal Cancer Subjects with KRAS p.G12C Mutation
CTID: null
Phase: Phase 3 Status: Restarted, Trial now transitioned, Ongoing
Date: 2022-01-26
GCAR-7213: GBM AGILE Global Adaptive Trial Master Protocol: An International, Seamless Phase II/III Response Adaptive Randomization Platform Trial Designed To Evaluate Multiple Regimens In Newly Diagnosed and Recurrent Glioblastoma (GBM), Version 3.2, Amendment 2.2, 14Apr2021
CTID: null
Phase: Phase 2, Phase 3 Status: Trial now transitioned, Completed
Date: 2021-12-07
A Phase 3 study of MK-4280A (coformulated favezelimab [MK-4280] plus
CTID: null
Phase: Phase 3 Status: Trial now transitioned, Temporarily Halted, Completed
Date: 2021-10-08
The Finnish National Study to Facilitate Patient Access to Targeted Anti-cancer Drugs to determine the Efficacy in Treatment of Advanced Cancers with a Known Molecular Profile
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2021-10-06
A randomized, phase IIb study of adjuvant durvalumab (MEDI4736)
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2021-09-27
Evaluation of treatment PERSOnalization based on its therapeutic monitoring in patients with metastatic colorectal cancer treated with regorafenib
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2021-04-27
A Phase 3 Randomized Study of Lenvatinib in Combination with Pembrolizumab Versus Standard of Care in Participants with Metastatic Colorectal Cancer Who Have Received and Progressed On or After or Became Intolerant to Prior Treatment
CTID: null
Phase: Phase 3 Status: Ongoing, Completed
Date: 2021-04-14
A Study Of Nivolumab In Combination With Trametinib With Or Without Ipilimumab In Participants With Previously Treated Metastatic Colorectal Cancers
CTID: null
Phase: Phase 1, Phase 2 Status: Restarted, Ongoing, Completed
Date: 2021-04-12
A phase II trial of neoadjuvant REGorafenib in combination with nIvolumab and short-course radiotherapy iN stage II-III rectAl cancer
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2021-03-09
REGOMAIN – A randomized, placebo-controlled, double-blinded, multicentre, comparative phase II study of the efficacy of regorafenib as maintenance treatment in patients with high grade bone sarcomas (HGBS) at diagnosis or relapse and without complete remission after standard treatment
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2021-01-07
A Multi-indication, Single-treatment Arm, Open-label Phase 2 Study of Regorafenib and Nivolumab in Combination with dose in Patients with Recurrent or Metastatic Solid Tumors
CTID: null
Phase: Phase 2 Status: Ongoing, GB - no longer in EU/EEA, Completed
Date: 2020-12-16
An Open-Label Study of Regorafenib in Combination with Pembrolizumab in Patients with Advanced or Metastatic Hepatocellular Carcinoma (HCC) after PD-1/PD-L1 Immune Checkpoint Inhibitors
CTID: null
Phase: Phase 2 Status: Temporarily Halted, Completed
Date: 2020-12-11
Randomized phase II study of PAnitumumab REchallenge followed by REgorafenib versus the reverse sequence in RAS and BRAF WILD-TYPE chemorefractory metastatic colorectal cancer patients.
CTID: null
Phase: Phase 2 Status: Trial now transitioned
Date: 2020-09-24
Regorafenib in combination with metronomic cyclophosphamide, capecitabine, and low-dose aspirin in metastatic colorectal cancer carcinoma
CTID: null
Phase: Phase 2 Status: Completed
Date: 2020-08-19
A randomized, phase II study comparing the sequences of regorafenib and trifluridine/tipiracil, after failure of standard therapies in patients with metastatic colorectal cancer
CTID: null
Phase: Phase 2 Status: Ongoing, Prematurely Ended
Date: 2020-07-23
A PHASE Ib/II, OPEN-LABEL, MULTICENTER, RANDOMIZED UMBRELLA STUDY EVALUATING THE EFFICACY AND SAFETY OF MULTIPLE IMMUNOTHERAPY-BASED TREATMENT COMBINATIONS IN PATIENTS WITH METASTATIC COLORECTAL CANCER (MORPHEUS-CRC)
CTID: null
Phase: Phase 1, Phase 2 Status: GB - no longer in EU/EEA, Prematurely Ended
Date: 2020-03-27
Gender-related response to Tyrosine Kinase-Inhibitor drugs in hepatocellular carcinoma
CTID: null
Phase: Phase 4 Status: Prematurely Ended
Date: 2020-03-20
The GOING Study: Regorafenib followed by Nivolumab in patients with Hepatocellular Carcinoma progressing under sorafenib
CTID: null
Phase: Phase 2 Status: Ongoing
Date: 2020-01-14
REGOSTA – A randomized, placebo-controlled, double-blinded, multicentre study evaluating the efficacy and safety of regorafenib as maintenance therapy after first-line treatment in patients with bone sarcomas
CTID: null
Phase: Phase 3 Status: Trial now transitioned
Date: 2019-03-14
A single arm, open-label, multicenter Phase 2 study of regorafenib in participants who have been treated in a previous Bayer-sponsored regorafenib study (monotherapy or combination treatment) that has reached the primary completion endpoint or the main data analysis, or has been stopped prematurely.
CTID: null
Phase: Phase 2 Status: GB - no longer in EU/EEA, Completed
Date: 2019-02-20
Safety, tolerability and efficacy of regorafenib in combination with FOLFIRINOX in patients with RAS-mutated metastatic colorectal cancer: a dose-escalation, phase I/II trial - FOLFIRINOX-R
CTID: null
Phase: Phase 1, Phase 2 Status: Ongoing
Date: 2018-12-04
A randomised phase II trial assessing REGorafenib combined with IRInotecan as second-line treatment in patients with metastatic gastro-oesophageal adenocarcinomas.
CTID: null
Phase: Phase 2 Status: Prematurely Ended
Date: 2018-11-09
Efficacy of regorafenib as maintenance therapy in non-adipocytic soft tissue sarcoma having received first-line doxorubicin-based chemotherapy
CTID: null
Phase: Phase 2 Status: Completed
Date: 2018-08-14
A randomized phase III trial assessing a regorafenib-irinotecan combination (REGIRI) versus regorafenib alone in metastatic colorectal cancer patients after failure of standard therapies, according to the A/A genotype of Cyclin D1
CTID: null
Phase: Phase 3 Status: Trial now transitioned
Date: 2018-08-06
An International, Multicenter, Open-label, Randomized, Phase 3 Study of BLU-285 vs Regorafenib in Patients with Locally Advanced Unresectable or Metastatic Gastrointestinal Stromal Tumor (GIST)
CTID: null
Phase: Phase 3 Status: GB - no longer in EU/EEA, Completed
Date: 2018-04-23
A phase I/II study of Regorafenib plus Avelumab in digestive tumors
CTID: null
Phase: Phase 1, Phase 2 Status: Ongoing
Date: 2018-03-14
A multi-center, open-label, non-randomized, phase I dose escalation study of regorafenib (BAY 73-4506) in pediatric subjects with solid malignant tumors that are recurrent or refractory to standard therapy.
CTID: null
Phase: Phase 1 Status: Ongoing, Completed
Date: 2018-01-15
Predictive value of in-vitro testing anti-cancer therapy sensitivity on tumorspheres from patients with metastatic colorectal cancer
CTID: null
Phase: Phase 2 Status: Completed
Date: 2017-07-10
Phase II randomized study of maintenance Regorafenib vs Placebo in no progression patients after first-line platinum and fluoropyrimidines based chemotherapy in HER2 negative locally advanced/metastatic gastric or gastroesophagel junction cancer (a-MANTRA Study)
CTID: null
Phase: Phase 2 Status: Completed
Date: 2017-04-04
A randomized phase II study between regorafenib and continuing biologic treatment to multi treated patients with colorectal cancer.
CTID: null
Phase: Phase 2 Status: Prematurely Ended
Date: 2016-09-07
A Phase III, open-label, multicenter, three-arm, randomized study to investigate the efficacy and safety of cobimetinib plus atezolizumab and atezolizumab monotherapy vs. regorafenib in patients with previously treated unresectable locally advanced or metastatic colorectal adenocarcinoma
CTID: null
Phase: Phase 3 Status: Prematurely Ended, Completed
Date: 2016-08-18
A randomized phase 2 study comparing different dose-approaches of induction treatment (first cycle) of regorafenib in metastatic colorectal cancer (mCRC) patients
CTID: null
Phase: Phase 2 Status: Completed
Date: 2016-06-09
The effects of the proton pump inhibitor esomeprazole on the bioavailability of regorafenib in patients with a metastatic colorectal cancer (mCRC) or gastrointestinal stromal tumour (GIST).
CTID: null
Phase: Phase 4 Status: Completed
Date: 2016-05-19
A randomised phase II trial of imatinib alternating with
CTID: null
Phase: Phase 2 Status: Ongoing, GB - no longer in EU/EEA, Prematurely Ended, Completed
Date: 2016-01-19
Phase II, single arm, non-randomized and multicenter clinical trial of regorafenib as a single agent in the first-line setting for patients with metastatic and/or unresectable KIT/PDGFR Wild Type GIST
CTID: null
Phase: Phase 2 Status: Ongoing, Completed
Date: 2015-10-13
PHASE II STUDY EVALUATION OF EFFICACITY AND TOLERANCE OF REGORAFENIB FOR 70 YEARS OLD AND MORE PATIENTS WITH A METASTATIC COLORECTAL ADENOCARCIMA
CTID: null
Phase: Phase 2 Status: Completed
Date: 2015-09-04
Phase II study on Regorafenib in advanced Solitary Fibrous Tumor
CTID: null
Phase: Phase 2 Status: Completed
Date: 2015-07-28
Regorafenib monotherapy as second-line treatment of patients with RAS-mutant advanced colorectal cancer: a multicentre, single-arm, two-stage, phase 2 study.
CTID: null
Phase: Phase 2 Status: Completed
Date: 2015-06-19
Molecular-biological tumor profiling for drug treatment selection in patients with advanced and refractory carcinoma
CTID: null
Phase: Phase 2 Status: Completed
Date: 2015-05-04
Regorafenib in relapsed glioblastoma. REGOMA study Randomized, controlled open‐label phase II clinical trial
CTID: null
Phase: Phase 2 Status: Completed
Date: 2015-04-24
A Randomized, Double-Blind Study of Ruxolitinib or Placebo in
CTID: null
Phase: Phase 2 Status: Completed, Prematurely Ended
Date: 2015-04-21
Phase II randomized study of maintenance regorafenib vs placebo in no progression patients after first-line platinum and fluoropyrimidines based chemotherapy in HER2 negative locally advanced/metastatic gastric or gastroesophagel junction cancer.
CTID: null
Phase: Phase 2 Status: Prematurely Ended
Date: 2014-12-24
REgorafenib’s Liquid BiopsY (RELY): A multicenter translational biomarker phase II trial of regorafenib in patients with non-resectable pretreated colorectal cancer.
CTID: null
Phase: Phase 2 Status: Completed
Date: 2014-12-17
Phase III study of RegorAfenib VErsus placebo as maintenance therapy in RAS wiLd type metastatic coLOrectal cancer
CTID: null
Phase: Phase 3 Status: Ongoing, Prematurely Ended
Date: 2014-12-01
An Open-Label Phase II Study of regorafenib In Patients With Metastatic Solid Tumors Who Have Progressed After Standard Therapy - RESOUND
CTID: null
Phase: Phase 2 Status: Ongoing
Date: 2014-11-25
A combined Phase IIa / IIb study of the efficacy, safety, and tolerability of repeated topical doses of regorafenib eye drops, in treatment-naïve subjects with neovascular age related macular degeneration
CTID: null
Phase: Phase 2 Status: Prematurely Ended
Date: 2014-07-02
Phase II study of Regorafenib as single agent for the treatment of patients with metastatic colorectal cancer (mCRC) with any RAS or BRAF mutation previously treated with FOLFOXIRI plus bevacizumab.
CTID: null
Phase: Phase 2 Status: Completed
Date: 2014-06-11
Activity of Regorafenib in combination with modified Gemcitabine - Oxaliplatin Chemotherapy (mGEMOX) in patients with advanced Biliary Tract Cancer (BTC): A Phase Ib-II trial
CTID: null
Phase: Phase 1, Phase 2 Status: Ongoing
Date: 2014-05-27
A Randomized Phase II, placebo-controlled , multicenter study evaluating efficacy and safety of regorafenib in patients with metastatic bone sarcomas.
CTID: null
Phase: Phase 2 Status: Trial now transitionelse if(down_display === 'none' || down_display === '') {
icon_angle_up.style.
































 463611831
463611831
