| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
Ornithine decarboxylase
|
|---|---|
| 体外研究 (In Vitro) |
依氟鸟氨酸是鸟氨酸脱羧酶的一种特异性、不可逆的抑制剂,被认为通过抑制毛囊中的这种酶来减缓头发生长[2]。
采用Franz扩散池对依氟鸟氨酸进行体外渗透研究。当将依氟鸟氨酸乳膏涂抹在用微针预处理过的小鼠皮肤区域时,依氟鸟氨酸的毛发生长抑制活性显著增强,很可能是因为微针产生的微孔允许依氟鸟氨酸渗透到皮肤中,这在体外渗透研究中得到了证实。免疫组织化学数据显示,将依氟鸟氨酸乳膏涂抹在微针预处理的皮肤区域,皮肤和毛囊的细胞增殖也明显受到抑制。[3] |
| 体内研究 (In Vivo) |
过去五十年来唯一被批准用于治疗人类非洲锥虫病的新药是依氟鸟氨酸。它主要用作对 melarsoprol 没有反应的布氏冈比亚锥虫感染的备用药物 [1]。当谈到减少毛发过多的参与者面部毛发的生长时,15% 依氟鸟氨酸霜的效果优于安慰剂。经过 24 周的治疗疗程后,58% 的依氟鸟氨酸患者和 34% 的安慰剂受试者的面部多毛症至少有所改善 [2]。当将依氟鸟氨酸乳膏施用到预先经过微针处理的小鼠皮肤区域时,依氟鸟氨酸的毛发生长抑制活性显着增加[3]。高血压14天后,依氟鸟氨酸治疗缩窄性高血压大鼠导致KCI和去甲肾上腺素收缩强度正常化以及乙酰胆碱松弛[4]。
|
| 酶活实验 |
In vitro permeation of eflornithine hydrochloride through mouse skin/盐酸依氟鸟氨酸体外经小鼠皮肤渗透研究[3]
如前所述,使用Franz扩散池仪完成体外渗透测定(Kumar等人,2012;Kumar et al. 2011;Naguib, Kumar, & Cui 2014)使用C57BL/6小鼠的下背皮肤。收集皮肤前24小时用电动剪发器修剪毛发。采集皮肤,用铝箔包裹,在- 20°C下保存最多一个月,并在需要时使用。在- 20°C下将皮肤冷冻(不使用冷冻保护剂)在文献中是常用的方法,并且这种皮肤样本经常用于渗透性研究(Stahl, Wohlert, & Kietzmann 2012)。Dennerlein等人表明,将新鲜切除的人体皮肤在- 20°C下冷冻和储存长达30天不会影响皮肤的渗透性(Dennerlein等人,2013年)。其他研究人员表明,将人体皮肤包裹在铝箔中并保存在- 26°C时,皮肤可保持其屏障性能长达6个月(Badran, Kuntsche, & Fahr 2009)。去除脂肪层后,将皮肤背朝上置于Franz扩散细胞上。接收器室装有5ml水,用Haake SC 100水循环器(ThermoScientific, Wellington, NH)保持在37°C。如前所述,在将毛发修饰的皮肤安装到Franz扩散池上之前,使用Dermaroller®微针辊进行处理(Kumar等人,2011;Naguib, Kumar, & Cui 2014)。将皮肤样品置于天平的平面上,微针滚轮在皮肤表面沿四个垂直方向滚动,每个方向滚动5次,共滚动20次,施加的压力为350 - 400g,在滚动滚轮的同时使用天平不断测量。皮肤扩散面积为0.64 cm2。供体室以500 μl的水注入盐酸依氟鸟氨酸4 mg,并用副膜覆盖以防止蒸发。在0、1、3、6、8和24 h后,从接收室中取出样品(150 μl),并立即补充新鲜培养基。使用高效液相色谱法对样品进行分析,方法采用先前描述的方法,并进行了修改(Saravanan et al. 2009)。色谱分析采用Agilent 1260 Infinity高效液相色谱仪,配备ZORBAX Eclipse Plus C18 (5 μm, 4.6 × 150 mm)色谱柱,流动相为乙腈-缓冲液(70%:30%,v/v)。将0.68 g磷酸一碱钾溶于1l水中制备缓冲液。流速0.8 ml/min。探测器波长为210 nm。 |
| 细胞实验 |
皮肤组织用福尔马林(10%)缓冲溶液固定24 h,用0.1 M磷酸钠缓冲液(pH 7.4)洗涤,用分级乙醇脱水,石蜡包埋,垂直切片。切片使用苏木精-伊红(H&E)或抗5-溴-2 ' -脱氧尿苷(BrdU)抗体在德克萨斯大学奥斯汀分校戴尔儿科研究所的组织学和组织处理设施进行染色。在安乐死前30分钟,以100 μg/g体重的剂量,在磷酸盐缓冲盐水(PBS, pH 7.4, 10 mM)中腹腔注射BrdU。所有皮肤切片均在Olympus BX53显微镜下检查[3]。
|
| 动物实验 |
本研究采用小鼠模型进行体内疗效研究,通过监测在预先用微针处理过的小鼠背部下方皮肤涂抹依氟鸟氨酸乳膏后毛发的再生情况来进行。同时,对治疗区域的皮肤和毛囊进行组织学检查[3]。
雌性C57BL/6小鼠(8-10周龄)由于其毛发生长周期与色素周期同步,是研究不同毛发生长周期阶段生理作用的理想模型(Slominski、Paus和Costantino,1991)。每个实验组包含3-4只小鼠。对麻醉小鼠背部下方皮肤的毛发进行以下处理:使用电动剃毛器修剪;使用之前描述的GiGi®蜂蜜热蜡拔毛(Xiao等,2012);或使用Nair®脱毛膏进行化学脱毛。去除毛发后的皮肤区域用刮匙涂抹13.9%盐酸依氟鸟氨酸乳膏(每次治疗每只小鼠约50毫克),每日两次,间隔至少8小时,最长持续36天。另一组小鼠用电推剪修剪了涂抹部位的毛发,并在每次涂抹依氟鸟氨酸乳膏前,按照先前描述的方法(Kumar等人,2012)使用微针滚轮进行治疗。简而言之,将小鼠放置在天平的平面上,用微针滚轮在标记的皮肤表面滚动10次,方向与小鼠体长平行,施加350-400克的压力(如天平所示)。对照组小鼠背部皮肤的毛发通过修剪、拔除或使用Nair®化学脱毛去除,但不使用依氟鸟氨酸乳膏进行治疗。在首次涂抹依氟鸟氨酸乳膏后,通过拍摄小鼠皮肤区域的数码照片,最长观察 36 天,来评估毛发再生情况。在研究的最后一天,对动物实施安乐死,并从治疗区域采集皮肤样本进行免疫组织化学研究。[3] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服依氟鸟氨酸后,依氟鸟氨酸的血浆峰浓度 (Cmax) 在给药后 3.5 小时达到峰值 (Tmax)。食物(高脂肪和高热量)不影响依氟鸟氨酸的 Cmax 和 AUC(浓度-时间曲线下面积)。将药片碾碎后加入标准布丁混合物中服用,对依氟鸟氨酸的暴露量(Cmax 和 AUC6h)没有影响。在临床使用条件下,对于面部多余毛发的女性,使用 13.9% (w/w) 的依氟鸟氨酸乳膏制剂,单次或多次给药后,其平均经皮吸收量低于放射性剂量的 1%。临床使用条件包括在放射性标记剂量给药前 2 小时内剃须,以及其他形式的剃毛、拔毛或镊子拔毛等去除面部毛发的方法。每日两次给药,四天内即可达到稳态。在临床使用条件下,对有面部多余毛发的女性(n=10)每天两次涂抹 0.5 克乳膏(总剂量 1.0 克/天;相当于 139 毫克无水盐酸依氟鸟氨酸),稳态 Cmax、Ctrough 和 AUC12hr 分别约为 10 ng/mL、5 ng/mL 和 92 ng hr/mL,以盐酸依氟鸟氨酸无水游离碱表示。在稳态下,每日两次涂抹0.5克乳膏(总剂量1.0克/天)后,依氟鸟氨酸的剂量标准化峰浓度(Cmax)和每日全身暴露量(AUC)预计分别比每日一次口服370毫克剂量低约100倍和60倍。 该化合物不发生代谢,主要以原形经尿液排出。 依氟鸟氨酸的分布容积(Vz/F)为24.3升。 依氟鸟氨酸的清除率(CL/F)为5.3升/小时。 在临床使用条件下,女性面部多毛症患者使用13.9%(w/w)乳膏制剂后,单次或多次给药后,依氟鸟氨酸的平均经皮吸收量低于放射性剂量的1%。临床使用条件包括在涂抹放射性标记药物前2小时内剃毛。除了其他形式的切割、拔毛或镊子拔毛等去除面部毛发的方法外, 在临床使用条件下,对有面部多余毛发的女性(n=10)每日两次涂抹0.5克乳膏(总剂量1.0克/天;相当于139毫克无水盐酸依氟鸟氨酸)后,以无水盐酸依氟鸟氨酸游离碱表示的稳态Cmax、Ctrough和AUC12hr分别约为10纳克/毫升、5纳克/毫升和92纳克/毫升。 在稳态下,与每日370毫克相比,每日两次涂抹0.5克乳膏(总剂量1.0克/天)后,依氟鸟氨酸的剂量标准化峰浓度(Cmax)和每日全身暴露量(AUC)估计分别降低约100倍和60倍。每日一次口服。 依氟鸟氨酸不经代谢,以原形经尿液排出。 有关依氟鸟氨酸(共8种代谢物)的更多吸收、分布和排泄(完整)数据,请访问HSDB记录页面。 代谢/代谢物 该化合物不经代谢,主要以原形经尿液排出。 生物半衰期 依氟鸟氨酸的终末血浆消除半衰期为3.5小时,表观稳态血浆半衰期约为8小时。 依氟鸟氨酸的表观稳态血浆t1/2约为8小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
妊娠期和哺乳期影响
◉ 哺乳期用药概述 母亲每日静脉注射依氟鸟氨酸 400 mg/kg,持续 7 天,未对母乳喂养的婴儿造成任何严重不良反应。外用依氟鸟氨酸吸收不良,因此不太可能进入婴儿血液循环,也不会对母乳喂养的婴儿造成任何不良反应。 ◉ 对母乳喂养婴儿的影响 在刚果民主共和国,一项针对 33 名婴儿的队列研究对服用硝呋莫司的住院母亲进行了母乳喂养(喂养程度未说明)进行了随访。其中 30 位母亲完成了 30 次口服硝呋莫司(15 mg/kg/天)的疗程,所有母亲均接受了 14 次静脉注射依氟鸟氨酸(400 mg/kg/天),持续 7 天,用于治疗人类非洲锥虫病(昏睡病)。哺乳期母亲平均同时服用4种其他药物,包括阿莫西林、环丙沙星、甲硝唑、复方磺胺甲噁唑、阿司匹林和双氯芬酸(各1例);氢化可的松、异丙嗪和奎宁(各2例);左旋咪唑(6例);磺胺多辛-乙胺嘧啶(8例);安乃近(13例);对乙酰氨基酚(16例);以及甲苯咪唑(17例)。所有母乳喂养的婴儿均未报告严重不良事件。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 依氟鸟氨酸不特异性结合人血浆蛋白。 |
| 参考文献 | |
| 其他信息 |
治疗用途
抗肿瘤药;酶抑制剂;锥虫杀灭剂 依氟鸟氨酸适用于减少女性面部多余毛发。依氟鸟氨酸仅在受影响个体的面部和下巴下方邻近受累区域进行过研究。使用应仅限于这些受累区域。/美国产品标签包含/ /实验性疗法:/ D,L-α-二氟甲基鸟氨酸 (DFMO) 于 20 多年前合成。人们曾希望这种酶激活的、不可逆的鸟氨酸脱羧酶抑制剂(鸟氨酸脱羧酶是多胺合成中的第一个酶)能够有效用于治疗增生性疾病,包括癌症和/或感染性疾病。研究发现,DFMO 通常对哺乳动物细胞和组织具有细胞抑制作用,其作为治疗药物的疗效有限。研究发现,高剂量DFMO会引起治疗限制性(但可逆性)耳毒性。这种副作用及其微弱的治疗活性导致许多临床医生对进一步开发DFMO作为癌症治疗药物失去了兴趣。然而,随后的研究表明,DFMO能够抑制多种啮齿动物模型中致癌物诱导的癌症发展,因此人们对开发该化合物作为预防剂的兴趣日益浓厚。近年来,抑制鸟氨酸脱羧酶作为癌症化学预防剂的理论依据得到了加强,因为研究表明,在某些细胞/组织类型中,该酶可被c-myc癌基因反式激活,并与ras癌基因协同作用,促进上皮组织的恶性转化。近期采用剂量递减设计的临床癌症化学预防试验表明,DFMO 可以长期低剂量给药,抑制胃肠道和其他上皮组织中的多胺含量,且不会引起可检测到的听力损失或其他副作用。目前正在进行的临床化学预防试验正在研究 DFMO 抑制特定上皮癌(包括结肠癌、食管癌、乳腺癌、皮肤癌和前列腺癌)高危人群中癌变替代终点生物标志物(例如结肠息肉复发)的疗效。 锥虫冈比亚型昏睡病的一线治疗药物通常是美拉胂醇,但如果治疗失败,预后通常不佳。多胺合成抑制剂依氟鸟氨酸(二氟甲基鸟氨酸,DFMO)已成为一种替代疗法。在扎伊尔农村地区,一项开放性试验采用三种不同的DFMO方案治疗了207例晚期冈比亚锥虫昏睡病患者。治疗期间,在DFMO给药前脑脊液中检测到锥虫的87例患者,其脑脊液中锥虫全部消失,脑脊液白细胞计数也从平均186/微升急剧下降至21/微升。152例患者在DFMO治疗后至少随访一年,仅13例(9%)复发。治疗失败在12岁以下儿童、仅接受口服DFMO治疗的患者以及将DFMO作为新确诊锥虫病初始治疗的患者中更为常见。毒性反应可接受。仅4例患者在治疗期间或治疗后不久死亡。骨髓抑制导致贫血(43%)或白细胞减少症(53%)较为常见,但影响甚微。这项开放性试验表明,DFMO 与美拉胂醇疗效相当,且毒性可能更低。由于经济和后勤方面的原因,DFMO 可能并非非洲农村地区的首选疗法,但对于绝大多数美拉胂醇治疗后复发的患者而言,DFMO 具有治愈作用。 ……一种多胺生物合成抑制剂依氟鸟氨酸(化学名称:DL-α-二氟甲基鸟氨酸,以盐酸盐一水合物形式供应)被用于治疗一名 3 岁半的儿童,该儿童新确诊患有严重的锥虫病,其感染病史为两年前在扎伊尔或刚果。治疗方案为:持续静脉输注依氟鸟氨酸 300 至 400 mg/kg/d,持续 25 天;随后口服依氟鸟氨酸 300 mg/kg/d,分四次等量服用,持续 17 天。患儿的康复情况显著,第一周内血液和脑脊液中的寄生虫即被清除。脑脊液细胞增多症完全消失。全身淋巴结肿大和发热症状逐渐消退。严重的共济失调、无法行走或自主改变姿势、明显的语言倒退以及嗜睡等症状在治疗期间和治疗后均有所改善。患儿对该药物耐受性良好;唯一观察到的不良反应是治疗第四周出现的短暂性血小板减少症。对于该患儿的中枢神经系统受累的锥虫病,依氟鸟氨酸是一种安全有效的治疗药物。 药物警告 禁忌症包括已知对依氟鸟氨酸或制剂中任何成分过敏。 如果出现过敏反应,应停止使用盐酸依氟鸟氨酸乳膏。 如果涂抹于破损或擦伤的皮肤上,可能会出现短暂的刺痛或灼烧感。易感患者或使用高于推荐剂量的患者可能会出现皮肤刺激。 目前尚未发现任何实验室检查异常与依氟鸟氨酸持续相关。在一项开放标签研究中,部分患者的转氨酶水平升高;然而,这些发现的临床意义尚不明确。 有关依氟鸟氨酸(共11项)的更多药物警告(完整)数据,请访问HSDB记录页面。 药效学 依氟鸟氨酸通过抑制多胺合成,降低MYCN扩增型神经母细胞瘤中致癌驱动基因MYCN和LIN28B的表达,从而恢复LIN28/Let-7代谢通路的平衡。该通路参与癌症干细胞和糖酵解代谢的调控。体外实验表明,依氟鸟氨酸可诱导MYCN扩增型和非扩增型神经母细胞瘤细胞衰老并抑制神经球形成,提示其具有细胞抑制作用。在注射了有限稀释度MYCN扩增的神经母细胞瘤细胞的小鼠中,用依氟鸟氨酸治疗可预防或延缓肿瘤的形成。此外,多胺也参与角蛋白的合成,抑制多胺可降低毛囊基质细胞的增殖,从而抑制毛发生长的生长期。 |
| 分子式 |
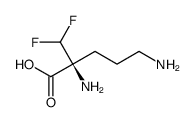
C6H12N2O2F2
|
|---|---|
| 分子量 |
182.16848
|
| 精确质量 |
182.087
|
| CAS号 |
67037-37-0
|
| 相关CAS号 |
Eflornithine;70052-12-9;L-Eflornithine monohydrochloride;69955-42-6;Eflornithine hydrochloride;68278-23-9;L-Eflornithine;66640-93-5; 96020-91-6 (HCl hydrate) 68278-23-9 (HCl); 70050-55-4 (R-isomer); 66640-93-5 (L-isomer); 69955-42-6 (S-isomer);
|
| PubChem CID |
3009
|
| 外观&性状 |
White to light yellow solid powder
|
| 密度 |
1.293g/cm3
|
| 沸点 |
347ºC at 760 mmHg
|
| 闪点 |
163.7ºC
|
| 折射率 |
1.462
|
| LogP |
1.173
|
| tPSA |
89.34
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
12
|
| 分子复杂度/Complexity |
166
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C(CC(C(F)F)(C(=O)O)N)CN
|
| InChi Key |
VLCYCQAOQCDTCN-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C6H12F2N2O2/c7-4(8)6(10,5(11)12)2-1-3-9/h4H,1-3,9-10H2,(H,11,12)
|
| 化学名 |
2,5-diamino-2-(difluoromethyl)pentanoic acid
|
| 别名 |
EFLORNITHINE; 70052-12-9; dfmo; Difluoromethylornithine; Ornidyl; 2-(Difluoromethyl)ornithine; 2,5-diamino-2-(difluoromethyl)pentanoic acid; ...; 67037-37-0;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
Typically soluble in DMSO (e.g. 10 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 5.4894 mL | 27.4469 mL | 54.8938 mL | |
| 5 mM | 1.0979 mL | 5.4894 mL | 10.9788 mL | |
| 10 mM | 0.5489 mL | 2.7447 mL | 5.4894 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT07287917
Conditions:Melanoma (Skin Cancer)|HER2-low Hormone Receptor Positive Breast CancerLink: https://clinicaltrials.gov/ct2/show/NCT04696029
Conditions:MedulloblastomaLink: https://clinicaltrials.gov/ct2/show/NCT02559778
Conditions:Neuroblastoma
Title:TArgeting Type 1 Diabetes Using POLyamines (TADPOL)
Status:Recruiting
updateDate:2026-04-20
Ctid:NCT05594563
Link: https://clinicaltrials.gov/ct2/show/NCT05594563
Conditions:Type 1 DiabetesLink: https://clinicaltrials.gov/ct2/show/NCT07468136
Conditions:Anaplastic Oligodendroglioma|Astrocytoma, IDH-Mutant, Grade 3|Astrocytoma, IDH-Mutant, Grade 4|Diffuse Astrocytoma|Glioblastoma, IDH-Wildtype|Malignant GliomaLink: https://clinicaltrials.gov/ct2/show/NCT06465199
Conditions:Atypical Teratoid/Rhabdoid Tumor|Embryonal Tumor With Multilayered Rosettes|Ewing Sarcoma|Diffuse Intrinsic Pontine Glioma|Osteosarcoma|Neuroblastoma|DIPG Brain TumorLink: https://clinicaltrials.gov/ct2/show/NCT06219174
Conditions:Non Small Cell Lung Cancer|Lung CancerLink: https://clinicaltrials.gov/ct2/show/NCT04301843
Conditions:NeuroblastomaLink: https://clinicaltrials.gov/ct2/show/NCT02679144
Conditions:NeuroblastomaLink: https://clinicaltrials.gov/ct2/show/NCT06059118
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT07321912
Conditions:Osteosarcoma|Ewing Sarcoma|Ewing Sarcoma MetastaticLink: https://clinicaltrials.gov/ct2/show/NCT05717153
Conditions:Diffuse Glioma|Malignant GliomaLink: https://clinicaltrials.gov/ct2/show/NCT06976424
Conditions:HirsuitismLink: https://clinicaltrials.gov/ct2/show/NCT05879367
Conditions:Glioblastoma, IDH-wildtype|Glioblastoma|Glioblastoma Multiforme|Glioblastoma IDH (Isocitrate Dehydrogenase) Wildtype|GBM|Astrocytoma|Astrocytoma, IDH-MutantLink: https://clinicaltrials.gov/ct2/show/NCT06892678
Conditions:Osteosarcoma Recurrent|Ewing's Tumor RecurrentLink: https://clinicaltrials.gov/ct2/show/NCT02030964
Conditions:NeuroblastomaLink: https://clinicaltrials.gov/ct2/show/NCT05500508
Conditions:Cancer|Solid Tumor|Solid Carcinoma|Advanced Cancer|DIPG Brain Tumor|Ovary Cancer|Breast Cancer|Papillary Thyroid Cancer|Head and Neck Cancer|Gastric Cancer|Nsclc|Mesotheliomas Pleural|Mesothelioma Peritoneum|Esophageal Cancer|Diffuse Midline Glioma, H3 K27M-Mutant|Endometrial Cancer|Cervical Cancer|Melanoma|Colorectal Cancer|Glioma, MalignantLink: https://clinicaltrials.gov/ct2/show/NCT02794428
Conditions:Gastric Cancer|Gastric Intestinal MetaplasiaLink: https://clinicaltrials.gov/ct2/show/NCT01059071
Conditions:NeuroblastomaLink: https://clinicaltrials.gov/ct2/show/NCT02395666
Conditions:NeuroblastomaLink: https://clinicaltrials.gov/ct2/show/NCT02139397
Conditions:Neuroblastoma RecurrentLink: https://clinicaltrials.gov/ct2/show/NCT04403282
Conditions:Pseudofolliculitis BarbaeLink: https://clinicaltrials.gov/ct2/show/NCT03536728
Conditions:Cancer|Solid Tumor|Solid Carcinoma|Advanced CancerLink: https://clinicaltrials.gov/ct2/show/NCT02796261
Conditions:Anaplastic Astrocytoma|Recurrent Anaplastic AstrocytomaLink: https://clinicaltrials.gov/ct2/show/NCT02384889
Conditions:Type 1 DiabetesLink: https://clinicaltrials.gov/ct2/show/NCT01483144
Conditions:Familial Adenomatous PolyposisLink: https://clinicaltrials.gov/ct2/show/NCT00033371
Conditions:Colorectal Cancer|Familial Adenomatous PolyposisLink: https://clinicaltrials.gov/ct2/show/NCT00204789
Conditions:Post-solid Organ Transplant|Skin NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT00005884
Conditions:Non-melanomatous Skin CancerLink: https://clinicaltrials.gov/ct2/show/NCT00983580
Conditions:Adenomatous PolypLink: https://clinicaltrials.gov/ct2/show/NCT00006079
Conditions:Cervical Cancer|Precancerous ConditionLink: https://clinicaltrials.gov/ct2/show/NCT00021294
Conditions:Non-melanomatous Skin Cancer|Precancerous/Nonmalignant ConditionLink: https://clinicaltrials.gov/ct2/show/NCT00006101
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT01685827
Conditions:Human African Trypanosomiasis (HAT)|Sleeping SicknessLink: https://clinicaltrials.gov/ct2/show/NCT00146627
Conditions:Trypanosomiasis, AfricanLink: https://clinicaltrials.gov/ct2/show/NCT00118365
Conditions:Precancerous ConditionLink: https://clinicaltrials.gov/ct2/show/NCT01636128
Conditions:Focus of Study: Drug Response Biomarkers, Chemoprevention, NeoplasmsLink: https://clinicaltrials.gov/ct2/show/NCT00003814
Conditions:Bladder CancerLink: https://clinicaltrials.gov/ct2/show/NCT00086736
Conditions:Prostate CancerLink: https://clinicaltrials.gov/ct2/show/NCT00003076
Conditions:Esophageal CancerLink: https://clinicaltrials.gov/ct2/show/NCT00330148
Conditions:Trypanosomiasis, African AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
